
|
STXBP1 Syndrom Interessante Links (Englisch): Breaking News: First Gene Therapy for STXBP1-RD |
|
STXBP1 Syndrom Interessante Links (Englisch): Breaking News: First Gene Therapy for STXBP1-RD |
 Datenschutz Impressum Kontakt Disclaimer |
Inhalt
|
2. Infografik STXBP1 Enzephalopathie 3. Elternbuch STXBP1 Enzephalopathie 13. Breaking News: Capsid-002 / Ravicti® 16. EU Register/Natural History Study und ESCO 17. STXBP1 Kinder in Deutschland |
Das Gen STXBP1
Seit 2008 wurden STXBP1- Mutationen bei Patienten festgestellt. Bei dem STXBP1 Syndrom treten die Genmutationen zu einem großen Anteil de novo auf; die Mutation ist nicht von einem der Eltern geerbt, sondern sie ist im betroffenen Individuum neu aufgetreten, ganz spontan in einer Keimzelle, Samenzelle oder Eizelle der Eltern, oder in der befruchteten Eizelle. Das STXBP1 Syndrom ist eine monogenetische Erkrankung des Gehirns, d.h. es ist nur ein einzelnes Gen defekt, welches auf einem Autosom liegt. Die Krankheit wird von einer autosomal-dominnate Genmutation ausgelöst; das auslösende Allel, d.h. wie ein Gen ein Merkmal ausprägt, ist nur einfach - auch heterozygot genannt - vorhanden. Heterozygot bedeutet, dass das Erbgut einer Zelle zwei unterschiedliche Kopien des STXBP1 Gens auf den beiden Chromosomen aufweist.
Der Mensch hat 23 Chromosom-Paare - also 46 Chromosomen:
- 2 Exemplaren von Chromosom 1, 2 Exemplaren von Chromosom 2, usw. bis Chromosom 22; das sind insgesamt 44 sogenannten Autosomen
- eine Frau hat zusätzlich 2 "X-Chromosomen" (Geschlechtschromosomen - d.h. 2 Gonosomen) und
- ein Mann hat zusätzlich ein "X-Chromosom" und ein "Y-Chromosom" (Geschlechtschromosomen - d.h. 2 Gonosomen)
Die Chromsomen bestehen aus bis zu 2 m lange DNA-Fäden, die an den Histonen und anderen Proteinen angelagert sind. Die 46 Chromosomen befinden sich in jeder Körperzelle.
Das STXBP1 Gen liegt auf dem langen Arm (q) des Chromosoms 9, Genlocus 34.11 und hat 20 Exons und erstreckt sich über 80.510 Basenparen. Exons sind DNA-Abschnitte eines Gens, die nach Transkription und Spleißen übrig bleiben und für Proteine codieren, z.B. Syntaxin-bindendes Protein 1. Die Basen Adenin (A), Cytosin (C), Guanin (G) und Thymin (T) sind vier Bausteine der DNA, die in einer genau festgelegten Reihenfolge zu einer langen Kette verknüpft sind. Diese Kette ist der Schlüssel für die Kodierung des Proteins. Eine DNA enthält etwa 20.000 Genen.
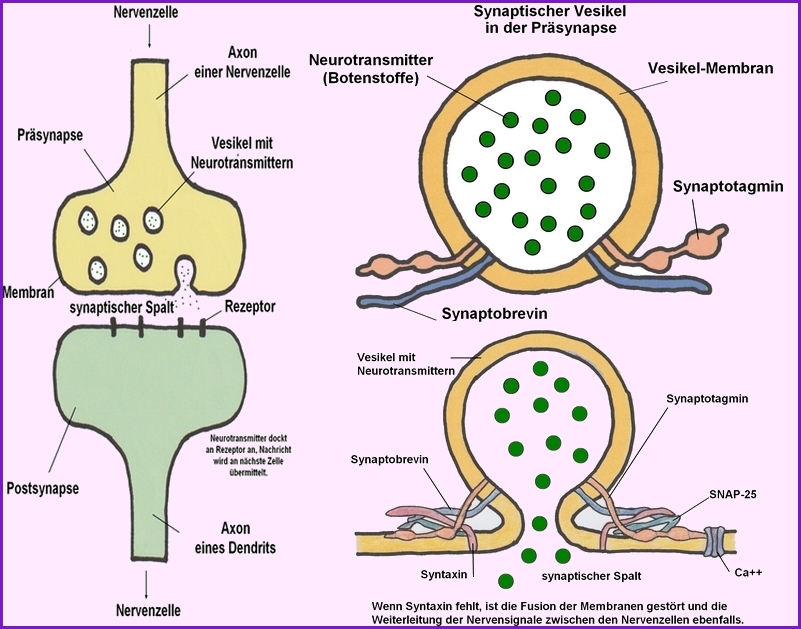
STXBP1 kodiert für das Syntaxin-bindende Protein 1, ein essenzielles Protein für die Regulation der Vesikelfreisetzung und Neurotransmittersekretion in den synaptischen Spalt:
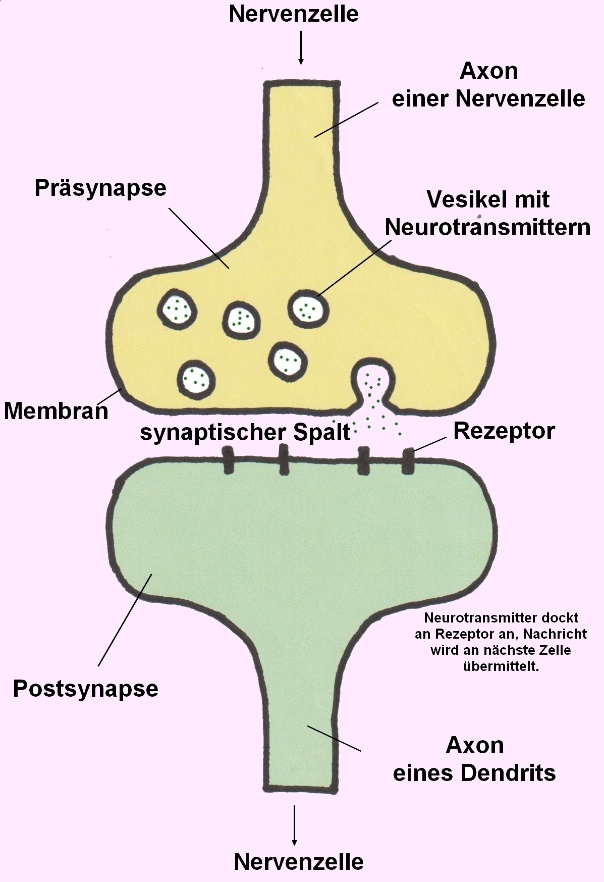
→ Prä- und Postsynapse von 2 Nervenzellen (Neuronen)- Bildnachweis: Gilberte Schnur
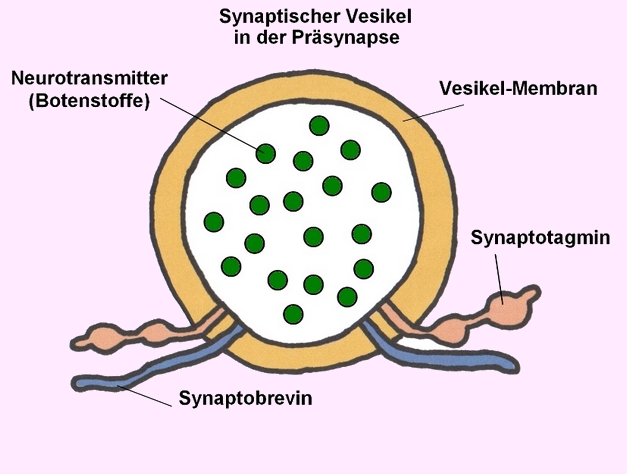
→ 1 Vesikel gefüllt mit Neurotransmittern (Botenstoffen) - Bildnachweis: Gilberte Schnur
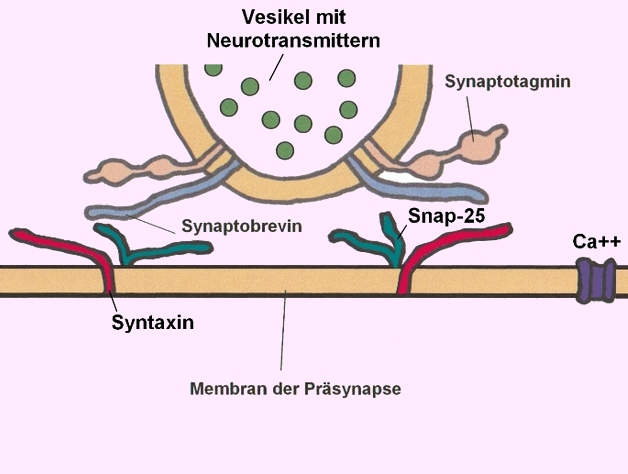
→ Membran der Präsynapse mit Syntaxin (rot) - Bildnachweis: Gilberte Schnur
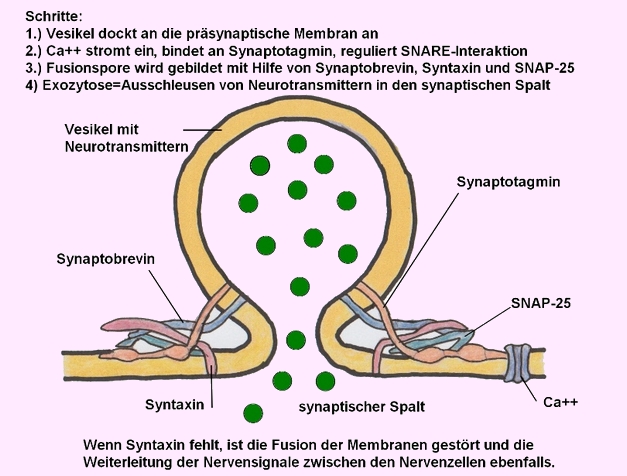
→ Ausschleusen von Neurotransmittern in den synaptischen Spalt - Funktion von Syntaxin - Bildnachweis: Gilberte Schnur
Mutationen
STXBP1 Mutationen sind Genmutationen im STXBP1 Gen selbst, sogenannte Punktmutationen. Diese können nur einzelne Basen oder auch größere DNA-Abschnitte betreffen. Bei einer Punktmutation ist nur ein Nucleotid verändert, oder nur ein Basenpaar gegen ein anderes ausgetauscht (Substitution).
Eine Substitution kann unterschiedliche Auswirkungen haben z.B.:
- stumme Mutation: es wird zwar durch den Austausch des Basenpaares ein anderes Codon gebildet, aber es wird jedoch in dieselbe Aminosäure übersetzt
- Missense-Mutation: der Austausch einer Aminosäure, deren Auswirkung auf die Funktionalität des Proteins von der Funktion des Proteins abhängt
- Nonsense-Mutation: es entsteht ein Stoppcodon, sodass die Translation vorzeitig abgebrochen wird; ein Basentriplett codiert durch Austausch eines Nucleotids zu einem Stopp-Codon
- Insertionen (Einfügen) oder Deletionen (Entfernen) einzelner Nucleotide: wenn sie im Exonbereich eines Gens auftreten, führen sie zu einer Verschiebung des Leserasters (Frameshift-Mutation), sodass ab der Läsion während der Translation die falschen Aminosäuren zur Synthese des Proteins verwendet werden.
Deletionen und Insertionen - sogenannte (Lese-)rastermutationen - beschränken sich nicht nur auf einzelne Basen, sondern können auch zwei, drei oder mehr Nucleotide umfassen.
Beispiel einer Frameshift-Mutation:
Eine Frameshift-Mutation wird durch Indels (Insertionen oder Deletionen) einer Anzahl von Nukleotiden in einer DNA-Sequenz verursacht, die nicht durch drei teilbar ist. Es verschiebt den Übersetzungsmechanismus von einem Leserahmen zu einem anderen. Nehmen wir an, wir haben eine Buchstabenfolge von THE FAT CAT SAT und der Buchstabe C wird gelöscht. Dann erhalten wir die Meldung THE FAT ATS AT ... Der Leserahmen ändert sich und das resultierende Protein funktioniert nicht richtig (oder überhaupt nicht).
STXBP1 Related Disorder - eine seltene Krankheit
Epilepsien des Kindes- und Jugendalters scheinen einen hohen genetischen Anteil zu haben, wie das auch bei dem STXBP1 Syndrom der Fall ist. Die epileptischen Enzephalopathien sind Erkrankungen bei denen eine früh einsetzende schwere Epilepsie, oft gekennzeichnet durch häufige tonische Anfälle oder Krämpfe ab dem Säuglingsalter, auftritt.
Symptome des STXBP1 Related Disorder
Die Krankheit geht mit einer Verlangsamung oder Rückbildung der Entwicklung und einer schweren kognitivsprachlichen Entwicklungsstörung, mit schweren Bewegungsauffälligkeiten und mentaler Retardierung und auch Verhaltensstörungen einher. Bei manchen Kindern zeigen sich auch Tremor und eine Störung der Grobmotorik / Koordination bzw. eine Ataxie auf. Bei manchen Kindern zeigt sich auch eine schlechte visuelle Verfolgung (CVI). Es werden aber auch STXBP1-Mutation diagnostiziert, die auch weniger schwere Epilepsie-Verlaufsformen zeigen oder bei denen die Kinder keine Epilepsie entwickeln.
Therapie?
Zur Zeit gibt es keine Heilung. Die Epilepsie ist bei betroffenen Kindern oft sehr schwer mit wirksamen Medikamenten einzustellen. Die Kinder haben viele Therapien wie Logo/Sprachtherapie, Physiotherapie, Ergotherapie und Sehfrüförderung.
Stxbp1 Forschungen haben angefangen, aber Forschung ist sehr teuer und auf Spenden angewiesen.
STXBP1 Infografik (Deutsch und Englisch) |
Im November 2024 wurde ein Informationsbuch für Eltern mit einem STXBP1 diagnostisierten Kind veröffentlicht. |
A. Deutsche Version STXBP1 e. V. - ein deutscher gemeinnütziger Verein |
B. English Version All STXBP1 kids need a cure. Therefore an association was founded in Germany. |
Was passiert im Gehirn?
Normalerweise sind elektrische und chemische Signale im Gehirn genau aufeinander abgestimmt und bestimmen die Tätigkeit der Nervenzellen im Gehirn. Wenn viele Nervenzellen sich zur gleichen Zeit in einzelnen Hirnregionen oder auch in beiden Gehirnhälften entladen, liegt eine Störung vor, und diese ungewohnten Impulse führen zu einem epileptischen Anfall. Epilepsie äußert sich in Form von epileptischen Anfällen.
Mittlerweile wurden eine zunehmende Anzahl an Genen als Ursache für Epilepsien entdeckt. Diese Genen kodieren für Ionenkanäle, die in der Zellmembran von Nervenzellen eingelagert sind. Diese sind am vesikulären synaptischen Transport beteiligt und beeinflussen die neuronale Erregung im Gehirn.


Credit Gilberte Schnur
Formen der Epilepsie
Im Allgemeinen werden epileptische Anfälle in zwei Hauptgruppen eingeteilt:
generalisierte Anfälle und fokale Anfälle.
Diese Einteilung hängt davon ab, wie und wo die Anfälle im Gehirn beginnen.
Generalisierte Anfälle sind von Spike-and-Wave-Entladungen im Gehirn gekennzeichnet; diese Entladungen breiten sich
von Anfang an auf beiden Seiten des Gehirns aus, und sie beginnen gleichzeitig auf der linken und rechten Gehirnhälfte.
Fokale Anfälle sind Entladungen, die nur an einem Ort im Gehirn, dem sogenannten Herd, stattfinden. Es gibt einfache fokale Anfälle (das Bewusstsein ist erhalten), komplexe fokale Anfälle (mit Bewusstseinsstörung) und fokale Anfälle mit Entwicklung zu sekundär generalisierten Anfällen.
Die Erscheinungsformen und Ausprägung der Epilepsie hängt also von der jeweils betroffenen Gehirnregion ab. Epileptische Anfälle sind nicht immer dramatisch und mit Verkrampfungen und Zuckungen verbunden, sondern können auch kaum merklich, wie bei kurzen Absenzen oder kurzen Missempfindungen ablaufen.
Führendes Zeichen bei Absenzen ist eine kurze "Abwesenheit" mit fehlender Ansprechbarkeit.
Ein großer Krampfanfall, auch mit Bewusstseinsverlust (Grand mal), der länger als 5 Minuten dauert, ist immer ein Notfall. Im Falle von mehr als fünf Minuten anhaltenden Anfällen, auch ohne Bewusstlosigkeit, liegt ein Status epilepticus vor. Ein Status epilepticus ist ein lebensbedrohlicher Notfall und muss sofort behandelt werden. Die Verabreichung eines Notfall-Medikaments ist dringend und eventuell auch die Einweisung in ein Krankenhaus.
Eine wichtige Untersuchung: das EEG
Durch eine Elektroenzephalographie (EEG) kann die Bereitschaft des Gehirns zu epileptischen Entladungen direkt angezeigt werden. Das EEG zeichnet die elektrische Aktivität des Gehirns auf und zeigt Muster einer normalen oder abnormalen elektrischen Aktivität im Gehirn. Das EEG-Gerät zeichnet die elektrische Aktivität des Gehirns als eine Reihe von Spuren auf. Jede Spur entspricht einer anderen Region des Gehirns. Sie können wie Spikes, scharfe Wellen und Spike-and-Wave-Entladungen aussehen. Bestimmte Muster weisen auf eine epileptische Bereitschaft hin. Das EEG zeigt auch in welchem Bereich des Gehirns die Anfälle auftreten.
Epileptische Enzephalopathien
Dies sind idiopathische, generalisierte und auch fokale Epilepsien. Krampfkranke Kinder zeigen oft abnormen ϑ-Rhythmen, Spikes und Waves im EEG. Die ϑ-Rhythmen zeigen sich vor allem bei den sogenannten frühkindlichen zentrencephalen Epilepsien wie z.B. bei astatischem und myoklonischem "Petit mal", bei frühkindlichen Absencen und bei zentrencephalem "Grand mal". Die ϑ-Rhythmen sind Symptom einer genetisch bedingten Krampfbereitschaft.
Das West-Syndrom
Beim West-Syndrom, auch noch Epilepsie mit Blitz-, Nick-, Salaam-Krämpfen (BNS-Epilepsie) genannt, handelt es sich um eine Epilepsie, die fast immer im Säuglingsalter mit Serien von 2 bis 150 kurzdauernden Anfällen beginnt. Das EEG zeigt ein typisches Hypsarrhythmie Muster.
Weitere Informationen zu Epilepsien finden Sie hier (auf Englisch): www.epilepsy.com
Zusätzliche Informationen zu der neuen Klassifikation der Epilepsien finden Sie hier (auf deutsch): Deutsche Gesellschaft für Epileptologie
Gelbe Liste Epilepsie
Therapie
Epilepsien sind oft schwere, oftmals therapieresistente Epilepsien, bei denen die epileptische Aktivität zu kognitiver Regression, Bewegungsstörungen oder psychomotorische Entwicklungsverzögerung führt.
Medikamentös ist die Epilepsie oft sehr schwer einzustellen. Die Behandlung besteht zunächst in der Gabe von anfallsunterdrückenden Medikamenten, die oft viele Nebenwirkungen haben. Auch andere Methoden wie die Epilepsiechirurgie können zum Einsatz in therapieresistenten Fällen kommen und zur Anfallsfreiheit führen. In diesem Fall muss das anfallsauslösende Areal exakt lokalisiert werden und die funktionelle Beeinträchtigung nach Entfernung des entsprechenden Hirngewebes abgeschätzt werden. Die Anfallsfrequenz kann ebenfalls durch eine Vagusnervstimulation (VNS) gesenkt werden.
Mehr Information über Epilepsie Forschung finden Sie hier (auf Englisch): Cure Epilepsy
Lesen Sie die Geschichten (auf Englisch) von Kindern, die mit Epilepsie zu kämpfen haben.
Klicken Sie hier: Caroline's Advocacy Journey
SUDEP - Plötzlicher Tod durch Epilepsie
Studie zur Sterblichkeit bei STXBP1 Patienten:
Early mortality in STXBP1-related disorders
Ein Thema worüber man nicht gerne spricht.
Dennoch, jede von Epilepsie betroffene oder auch nur gefährdete Person sollte über den Plötzlichen Epilepsietod (SUDEP) unbedingt Bescheid wissen. Denn es ist ein Risiko, dass nicht zu unterschätzen ist.
Einer von 1.000 Epilepsiepatienten verstirbt jedes Jahr an SUDEP. Und das obwohl viele Fälle vermeidbar sind.
Epilepsie ist die weltweit am häufigsten auftretende Krankheit, die weder Alter noch Rasse, soziale Schicht, Klasse, natürliche oder geografische Grenzen kennt. In jedem Lebensalter können epileptische Anfälle, auch als einzelner Anfall, auftreten.
Von Epilepsie spricht man aber erst, wenn Anfälle sich wiederholen. Wenn das Gehirn oder auch einzelne Hirnbereiche übermäßig aktiv sind und zu viele Signale senden, ist das Gleichgewicht im Gehirn verstört und es entstehen epileptische Anfälle.
SUDEP steht für "Sudden Unexpected Death in Epilepsy" und beschreibt den plötzlichen Tod bei einem Epilepsiepatienten, der als Folge eines Anfalls aus einem weitgehend normalen Gesundheitszustand ohne weitere erkennbare Ursachen plötzlich verstirbt.
SUDEP wird in den meisten Fällen durch den Zusammenbruch des Herz-Lungen-Kreislaufs Systems verursacht. Bei einem epileptischen Anfall kann es kurz zu Funktionsausfällen der Regulierung von Atmung (Apnea) und Herzschlag (in Form von sehr hoher Frequenz oder Verlangsamung) im Gehirn kommen, was zu einem Atem- und Herzstillstand führen kann.
Deshalb ist es wichtig über SUDEP Bescheid zu wissen, denn durch rechtzeitige Erste-Hilfe-Maßnahmen kann unterstützt werden, das die Atmung wieder einsetzt. Dazu reichen manchmal schon ganz einfache Maßnahmen, wie Berührung und Ansprechen. Ein snelles Handeln ist hierbei wichtig, denn die größsten Chancen, auf ein erfolgreiches Eingreifen bestehen in den ersten 3 Minuten nach dem Aussetzen der Herz-Lungen Funktion.
Die meisten SUDEP Fälle geschehen im Schlaf während eines epileptischen Anfalls oder unmittelbar danach, häufig in den frühen Morgenstunden.
Besonders risikoreich ist die dramatischste Form epileptischer Anfälle: die generalisierten tonisch-klonischen Anfälle (TKA), auch Grand mal genannt. Bei diesen Anfällen kann es in der postiktalen Phase (Zustand unmittelbar nach dem TKA Anfall) zu einem Herz-Kreislauf-Zusammenbruch kommen.
Deshalb ist Vorsicht geboten und es gibt Möglichkeiten einen SUDEP zu verhindern:
- regelmäßige Medikamenten Einnahme bei Epilepsie; das Erreichen einer vollständigen Anfallsfreiheit mit Medikamenten
- regelmäßiger Nachtschlaf und Schlafen in Rückenposition
- nächtliche Überwachung mit klinisch geprüften Geräten, die durch Warnungen bei einem gefährlichen Herzrhythmus oder einem Herzstillstand oder bei Atmungspausen oder Atmungsstillstand, wodurch der Sauerstoffgehalt auf ein lebensbedrohliches Niveau sinkt, rechtzeitige Notfallmaßnahmen ermöglichen.
Um eine bessere Kontrolle der nächtlichen Anfälle zu erreichen werden sensortechnischen Überwachungsgeräten zur nächtlichen Anfallsdetektion empfohlen.
Solche Geräte können eine gefährliche Situation erkennen, indem das Gerät einen TKA oder sonstigen motorischen Anfall anzeigt, das Abfallen der Sauerstoffsättigung des Bluts anzeigt, Veränderungen des Herzrhythmus oder Pulses angibt, oder auch sonstige physiologische Parameter in Form eines Alarms meldet.
Auch ist es wichtig sich regelmäßig in Erster Hilfe / Laienreanimation schulen zu lassen, damit man für den Notfall vorbereitet ist.
Es ist wichtig nach einem Anfall darauf zu achten, dass die Atmung wieder einsetzt. Es wird empfohlen, die Atmung 45 bis 60 Minuten zu beobachten. Bei einem Notfall sind die ersten 3 Minuten entscheidend.
(Quelle: SUDEP.de - Oskar Killinger Stiftung)
Was sagt der genetische Befund aus?
Der Gentest
Gentests dienen dem Nachweis von Veränderungen an Chromosomen, Genen oder Proteinen.
Die Ergebnisse können bestätigen oder ausschließen, dass das medizinische Problem einer Person durch ein fehlerhaftes Gen verursacht wird.
Bestehende Technologien haben sich in den letzten Jahren zu neuen Möglichkeiten in der Humangenomforschung entwickelt.
Die neuen Methoden werden unter dem Begriff Next Generation Sequencing (NGS) zusammengefasst.
Diese beinhalten z. B.
Jedes Gewebe, das kernhaltige Zellen mit der DNA enthält, kann zur Bestimmung einer Mutation verwendet werden - zum Beispiel Blut, Sperma, Haar, Knochen, Haut oder Speichel.
Bei der Untersuchung eines einzelnen Gens, einer Gruppe von Genen oder des gesamten Genoms wird die DNA-Sequenz des Kindes mit einer Referenzsequenz verglichen, um festzustellen, ob es Unterschiede gibt.
Eine Referenzsequenz (auch Wildtyp genannt) ist eine DNA-Sequenz, die nicht mit einer Krankheit in Verbindung gebracht wird. Jeder Unterschied in der DNA-Sequenz des Kindes im Vergleich zur Referenzsequenz wird als Variante bezeichnet.
Die Technologien
Es werden verschiedene Technologien verwendet:
Die Isoformen des STXBP1-Gens
Es gibt zwei Isoformen des STXBP1-Gens:
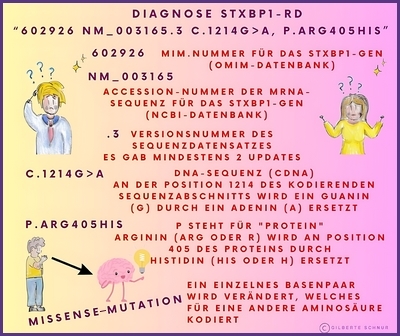
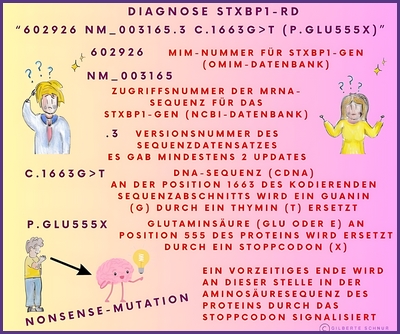
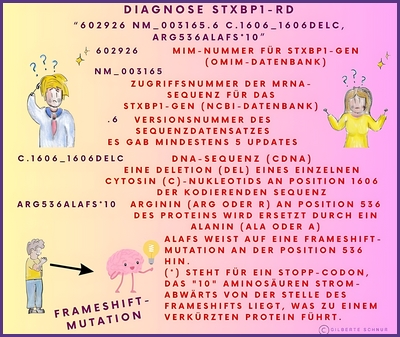
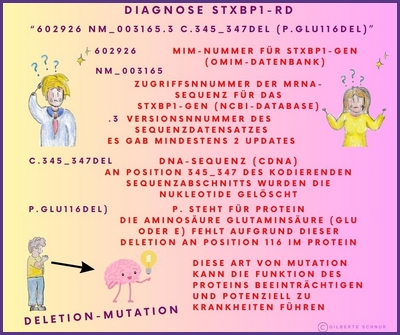
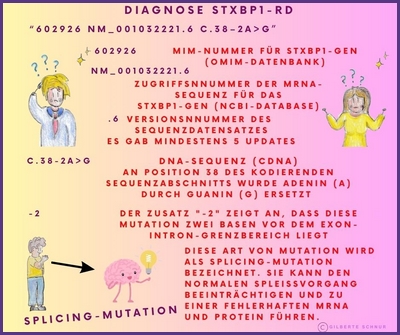
In einem genetischen Bericht wird die Referenzsequenz oft in Form eines alphanumerischen Codes wie NM_003165.6 oder auch NM_003165.3, der Zahl 6 oder 3 (auch eine andere ist möglich) verwendet. Dies liegt daran, dass die Ergebnisse der genetischen Sequenzierung eines Gens in GenBank (DNA-Sequenzdatenbank) als Teilsequenzen eingereicht werden können, die im Laufe der Zeit von verschiedenen Forschern identifiziert wurden.
Was steht in einem Gentestbericht?
In dem Gentestbericht werden einzelne Varianten, die durch Gentests in der DNA eines Kindes gefunden werden, beschrieben.
Im Falle der STXBP1-Krankheit würde ein Gentest eine "pathogene Variante" (pathogen=krankheitsverursachend) oder "Mutation im STXBP1-Gen" nachweisen.
Die Arten von Varianten (Mutationen), die in einem Gentestbericht angegeben werden, werden mit einer Art Kurzschrift (einem alphanumerischen Code) beschrieben, die die Veränderung in der DNA-Sequenz und die entsprechende Veränderung im Protein, aus dem das Gen besteht, beschreibt.
Hier einige Beispiele dieser Kurzschriften in Bildern erklärt:

Außergewöhnliche Ereignisse für STXBP1 Forscher und Familien
Auf der diesjährige Jahrestagung der American Epilepsy Society (AES) im Dezember 2024 kamen über 5.000 Teilnehmer, wie z. B. Epilepsie-Experten zusammen.
Auf der Jahrestagung der AES treffen sich Gesundheitsdienstleister, Wissenschaftler, Interessenvertreter, Vertreter der Industrie und andere Fachleute, die sich für bessere Ergebnisse für Menschen mit Epilepsie einsetzen. Die Teilnehmer freuten sich im Jahr 2024 auf über 120 Sitzungen.
Auch wichtige STXBP1-Highlights waren mehrere Projekte. Es gab Vorträge und 18 Poster- und Vortragspräsentationen, in denen STXBP1 erwähnt wurde, u.a.:
- die Daten der STARR-Studie wurde in mehreren Postern vorgestellt
- das Team des Children's Hospital of Philadelphia (CHOP) unter der Leitung von Ingo Helbig stellte drei Poster vor
- Ben Prosser von UPen sprach über die Entwicklung von ASOs für STXBP1 und Syngap1 durch sein Labor
- Peter Galer stellte seine Ergebnisse zur Analyse STXBP1-spezigischer EEG-Muster vor, vielleicht wichtige Biomaker für STXBP1
- Capside Biotherapeutics beschrieb ihre Gentherapie für STXBP1 und die Fähigkeit ihres viralen Vektors zur Expression des STXBP1-Gens in nichtmenschlichen Primaten
Zusammenfassung AES 2024
Capsida: CAP-002
Ein Blick auf AES25
Am 20. Oktober 2023 organisierte die STXBP1-Stiftung ein erstes Externally-Led Patient-Focused Drug Development Meeting (EL-PFDD)mit der FDA, um die Perspektive von Patienten mit STXBP1-bezogenen Erkrankungen vorzustellen. Ziel war es, die FDA und andere Interessengruppen über die Herausforderungen, mit denen STXBP1-Patienten und Familien leben, zu informieren.
Weitere Infos auf der Webseite der STXBP1-Stiftung:
STXBP1 Meeting mit der FDA
Wieviele Menschen auf der Welt haben eine STXBP1 Diagnose?
Global Connect
Die STXBP1 Gemeinschaft hat ein globales Elternnetzwerk, das sich aus vielen Gruppen aus der ganzen Welt zusammensetzt gebildet. Es setzt sich dafür ein, das Leben von STXBP1-Patienten zu verbessern.
Das Netzwerk besteht schon aus vielen Teilnehmern : Global Connect
EIN PAAR AUFREGENDE HIGHLIGHTS DER ZEHNTEN ZÄHLUNG:

- 1378 Patienten weltweit (Stand 29. Juni 2025)
- 44 % in Europa, 32 % in Nordamerika, 16 % im asiatisch-pazifischen Raum, 7 % Rest der Welt
- Die Altersdaten sind in den Ländern, für die wir sie haben, ziemlich einheitlich. Die beiden größten Gruppen sind 6 bis 12 Jahre alt (34 %) und 0 bis 5 Jahre alt (30 %). Bemerkenswert ist auch, dass wir 162 erwachsene STX-Mitglieder (ab 18 Jahren und älter) haben
- Die Geschlechterdaten sind gleichmäßig verteilt: 49% Frauen, 51% Männer.
(Alters- und Geschlechtsdaten sind für 75 % der Personen verfügbar)
In Deutschland gibt es 87 STXer.
(Credit Daten und Grafik: STXBP1disorders.org)
EIN PAAR AUFREGENDE HIGHLIGHTS DER NEUNTEN ZÄHLUNG:

- 1310 Patienten weltweit (Stand 30. Mä 2025)
- 44 % in Europa, 32 % in Nordamerika, 16 % im asiatisch-pazifischen Raum, 7 % Rest der Welt
- Die Altersdaten sind in den Ländern, für die wir sie haben, ziemlich einheitlich. Die beiden größten Gruppen sind 6 bis 12 Jahre alt (34 %) und 0 bis 5 Jahre alt (30 %). Bemerkenswert ist auch, dass wir 158 erwachsene STX-Mitglieder (ab 18 Jahren und älter) haben
- Die Geschlechterdaten sind gleichmäßig verteilt: 50% Frauen, 50% Männer.
(Alters- und Geschlechtsdaten sind für 82 % der Personen verfügbar)
In Deutschland gibt es 84 STXer.
(Credit Daten und Grafik: STXBP1disorders.org)
EIN PAAR AUFREGENDE HIGHLIGHTS DER ACHTEN ZÄHLUNG:

- 1227 Patienten weltweit (Stand 3. Januar 2025)
- 43 % in Europa, 32 % in Nordamerika, 18 % im asiatisch-pazifischen Raum, 7 % Rest der Welt
- Die Altersdaten sind in den Ländern, für die wir sie haben, ziemlich einheitlich. Die beiden größten Gruppen sind 6 bis 12 Jahre alt (34 %) und 0 bis 5 Jahre alt (30 %). Bemerkenswert ist auch, dass wir 155 erwachsene STX-Mitglieder (ab 18 Jahren und älter) haben
- Die Geschlechterdaten sind gleichmäßig verteilt: 50% Frauen, 50% Männer.
(Alters- und Geschlechtsdaten sind für 78 % der Personen verfügbar)
In Deutschland gibt es 84 STXer.
(Credit Daten und Grafik: STXBP1disorders.org)
EIN PAAR AUFREGENDE HIGHLIGHTS DER SIEBTEN ZÄHLUNG:

- 1157 Patienten weltweit (Stand 30. September 2024)
- 44 % in Europa, 32 % in Nordamerika, 16 % im asiatisch-pazifischen Raum, 8 % Rest der Welt
- Die Altersdaten sind in den Ländern, für die wir sie haben, ziemlich einheitlich. Die beiden größten Gruppen sind 6 bis 12 Jahre alt (34 %) und 0 bis 5 Jahre alt (30 %). Bemerkenswert ist auch, dass wir 140 erwachsene STX-Mitglieder (ab 18 Jahren und älter) haben
- Die Geschlechterdaten sind gleichmäßig verteilt: 50% Frauen, 50% Männer.
(Alters- und Geschlechtsdaten sind für 77 % der Personen verfügbar)
In Deutschland gibt es 83 STXer.
(Credit Daten und Grafik: STXBP1disorders.org)
EIN PAAR AUFREGENDE HIGHLIGHTS DER SECHSTEN ZÄHLUNG:

- 1124 Patienten weltweit (Stand 3. Juli 2024)
- 44 % in Europa, 32 % in Nordamerika, 16 % im asiatisch-pazifischen Raum, 8 % Rest der Welt
- Die Altersdaten sind in den Ländern, für die wir sie haben, ziemlich einheitlich. Die beiden größten Gruppen sind 6 bis 12 Jahre alt (34 %) und 0 bis 5 Jahre alt (30 %). Bemerkenswert ist auch, dass wir 136 erwachsene STX-Mitglieder (ab 18 Jahren und älter) haben
- Die Geschlechterdaten sind gleichmäßig verteilt: 50% Frauen, 50% Männer.
(Alters- und Geschlechtsdaten sind für 76 % der Personen verfügbar)
In Deutschland gibt es 81 STXer.
(Credit Daten und Grafik: STXBP1disorders.org)
EIN PAAR AUFREGENDE HIGHLIGHTS DER FÜNFTEN ZÄHLUNG:

- 1085 Patienten weltweit (Stand 30. März 2024)
- 43 % in Europa, 32 % in Nordamerika, 16 % im asiatisch-pazifischen Raum, 9 % Rest der Welt
- Die Altersdaten sind in den Ländern, für die wir sie haben, ziemlich einheitlich. Die beiden größten Gruppen sind 6 bis 12 Jahre alt (34 %) und 0 bis 5 Jahre alt (30 %). Bemerkenswert ist auch, dass wir 131 erwachsene STX-Mitglieder (ab 18 Jahren und älter) haben
- Die Geschlechterdaten sind gleichmäßig verteilt: 50% Frauen, 50% Männer.
(Alters- und Geschlechtsdaten sind für 76 % der Personen verfügbar)
In Deutschland gibt es 78 STXer.
(Credit Daten und Grafik: STXBP1disorders.org)
EIN PAAR AUFREGENDE HIGHLIGHTS DER VIERTEN ZÄHLUNG:

- 973 Patienten weltweit (Stand 30. Dezember 2023)
- 42 % in Europa, 34 % in Nordamerika, 16 % im asiatisch-pazifischen Raum, 8 % Rest der Welt
- Die Altersdaten sind in den Ländern, für die wir sie haben, ziemlich einheitlich. Die beiden größten Gruppen sind 6 bis 12 Jahre alt (36 %) und 0 bis 5 Jahre alt (34 %). Bemerkenswert ist auch, dass wir 111 erwachsene STX-Mitglieder (ab 18 Jahren und älter) haben
- Die Geschlechterdaten sind gleichmäßig verteilt: 50% Frauen, 50% Männer.
(Alters- und Geschlechtsdaten sind für 78 % der Personen verfügbar)
In Deutschland gibt es 74 STXer.
(Credit Daten und Grafik: STXBP1disorders.org)
EIN PAAR AUFREGENDE HIGHLIGHTS DER DRITTEN ZÄHLUNG:

- 951 Patienten weltweit (Stand 31. September 2023)
- 42 % in Europa, 34 % in Nordamerika, 16 % im asiatisch-pazifischen Raum, 8 % Rest der Welt
- Die Altersdaten sind in den Ländern, für die wir sie haben, ziemlich einheitlich. Die beiden größten Gruppen sind 6 bis 12 Jahre alt (36 %) und 0 bis 5 Jahre alt (34 %). Bemerkenswert ist auch, dass wir 110 erwachsene STX-Mitglieder (ab 18 Jahren und älter) haben
- Die Geschlechterdaten sind gleichmäßig verteilt: 49% Frauen, 51% Männer.
(Alters- und Geschlechtsdaten sind für 78 % der Personen verfügbar)
In Deutschland gibt es 72 STXler.
(Credit Daten und Grafik: STXBP1disorders.org)
EIN PAAR AUFREGENDE HIGHLIGHTS DER ZWEITEN ZÄHLUNG:

- 928 Patienten weltweit (Stand 30. Juni 2023)
- 42 % in Europa, 34 % in Nordamerika, 16 % im asiatisch-pazifischen Raum, 8 % Rest der Welt
- Die Altersdaten sind in den Ländern, für die wir sie haben, ziemlich konsistent. Die beiden größten Gruppen sind 6 bis 12 Jahre alt (36 %) und 0 bis 5 Jahre alt (35 %). Bemerkenswert ist auch, dass wir 104 erwachsene STX-Mitglieder (ab 18 Jahren) haben
- Die Geschlechterdaten sind gleichmäßig verteilt: 49% Frauen, 51% Männer.
(Alters- und Geschlechtsdaten sind für 80 % der Personen verfügbar)
In Deutschland gibt es 71 STXler.
(Credit Daten und Grafik: STXBP1disorders.org)
EIN PAAR AUFREGENDE HIGHLIGHTS DER ERSTEN ZÄHLUNG:

- 903 Patienten weltweit (Stand 31. März 2023)
- 42 % in Europa, 35 % in Nordamerika, 16 % im asiatisch-pazifischen Raum, 7 % Rest der Welt
- Die Altersdaten sind in den Ländern ziemlich einheitlich. Die beiden größten Gruppen sind 6 bis 12 Jahre alt (36 %) und 0 bis 5 Jahre alt (34 %)
- Die Geschlechtsdaten sind gleichmäßig aufgeteilt: 49% Frauen, 51% Männer.
(Alters- und Geschlechtsangaben liegen für 76 % der Personen vor)
Bemerkenswert ist auch, dass es 99 erwachsene STX'er gibt (18 und älter).
In Deutschland gibt es 67 STXler.
(Credit Daten und Grafik: STXBP1disorders.org)
Heidelberg 8. - 11. Oktober 2025
Vom 8. bis 11. Oktober 2025 treffen sich Familien, Kliniker und Forscher in Deutschland. Der STXBP1-Gipfel 2025 wird vom European STXBP1 Consortium (ESCO) ausgerichtet.
Die Forscher treffen sich vom 8. bis 10. Oktober, um Fortschritte zu diskutieren, Wissen auszutauschen und die Zusammenarbeit zu fördern.
Registrierung hier:
Anmeldung für:
Akademische Forscher, Doktoranden, Kliniker, Vertreter der Industrie
Anmeldung für:
Familien
Am 11. Oktober findet der Familientag statt, an dem die Eltern sich kennenlernen und austauschen können.


Credit Fotos ESCO
Weitere Infos auf STXBP1-Gipfel 2025 in Heidelberg
Summit 2025 in USA
STXBP1 Foundation organisierte ein Summit vom 18. bis zum 20. Juli in Westminster, CO (außerhalb von Denver).
Der STXBP1 Summit+ Family Meeting war voller Informationen, Forschungsergebnisse, Kontakte, Emotionen, Spaß, Auszeichnungen und einfach: Was für ein Wochenende!
Die gesamten 8 Stunden des Samstags, 19. Juli 2025, und die einzelnen Sitzungen wurden aufgezeichnet und der Link hierzu befindet sich auf der STXBP1 Foundation Webseite:
STXBP1-Summit 2025 in Colorado
Drexelbrook Event Center in Drexel Hill, PA, USA vom 18. - 21. Juli 2024
Vom 18. bis 21. Juli 2024 trafen sich Familien, Kliniker und Forscher in Drexelbrook Event Center in Drexel Hill, PA. Die Veranstaltung fand statt, um die neuesten Ergebnissen zu der STXBP1-Erkrankung und Forschung zu erfahren und zu besprechen.
Die Videos der Veranstaltung vom 18. und 19. Juli 2024 können hier angeschaut werden:
Videos vom 18. und 19. Juli 2024
Hier das Treffen im Überblick:
Freitag und Samstag, 18.7. - 19.7.: STXBP1-Kliniktage - meeting mit Forschern
Samstag und Sonntag 20.7. und 21.7.: Familientage
Information finden Sie auf der Webseite der STXBP1 Foundation:
Research Roundtable Agenda:
Forscher-Meeting Summit+ 2024
Familientag 1: Wissenschaftliche Sitzungen und Familientag 2: Unterstützungs-Sitzungen
Familientage Summit+ 2024
Milan 16. - 18. Mai 2023
Vom 16. bis 18. Mai 2023 trafen sich Familien, Kliniker und Forscher zum ersten Mal in Europa, in Italien, Milan.
Die Videos der Präsentationen in Milan sind nun online:
STXBP1 Summit 2023 in Milan - Videos (English)
Credit Videos: STXBP1 Foundation
Hier finden Sie die deutsche Präsentation, die am 3. Tag in Milan gezeigt wurde:
Video Copyright Gilberte Schnur
Deutsche Eltern danken Forschern und allen, die sich für die STXBP1 Gemeinschaft einsetzen.

Credit Fotos Eltern der Whatsapp Gruppe
Mit diesem ersten europäischen STXBP1-Gipfel will die STXBP1 Gemeinschaft Fachleute aus dem Gesundheitswesen und Grundlagenforscher, die sich mit STXBP1 beschäftigen,
mit Patienten und ihren Betreuern zusammenbringen. Das Ziel der Veranstaltung war Kooperationsnetzwerke zu fördern.
Forscher aus der ganzen Welt tauschten Wissen aus und
in Europa soll das Bewusstsein für STXBP1-E geschärft werden.
Nur durch eine umfassende Zusammenarbeit, sowohl mit der internationalen Forschungsgemeinschaft als auch mit den STXBP1-Familien, ist die Suche nach einer Heilung möglich.
Hier das Treffen im Überblick:
Dienstag, 16.5.: STXBP1 Forscher Meeting; STXBP1 im Labor
Mittwoch, 17.5.: STXBP1 Forscher Meeting; STXBP1 in der Klinik
Donnerstag, 18.5.: STXBP1 Familientag - Meeting mit Forschern
Das Programm der Fakultät und der wissenschaftlichen Sitzungen stand als pdf zur Verfügung.
STXBP1 Summit 2023 in Milan - Programm
Weitere Infos zu den einzelnen Tagen finden Sie auf dieser Webseite:
CNCR co-hosts 1st European STXBP1 Summit in Milan
Philadelphia 19. - 20. August 2022
Vom 19. bis 20. August 2022 trafen sich Familien, Kliniker und Forscher in Philadelphia, um die neuesten Ergebnissen zu der STXBP1-Erkrankung und Forschung zu erfahren und zu besprechen.
Sie können alle Vorträge auf der Webseite der STXBP1 Foundation ansehen:
Vidoes STXBP1 Summit+ 2022
Abonnieren Sie auch den YouTube-Kanal der STXBP1 Foundation, um Video der STXBP1 Foundation anzuschauen.
Das Programm können Sie als pdf hier downloaden: Programm STXBP1 Summit+
Die Ergebnisse der Natural History Study in Deutschland wurden auf dem STXBP1 Summit+ vorgestellt und auf dieser Webseite unter dem Button "Forschung DE" veröffentlicht.
STXBP1 Awareness Month 2025
Im September 2025 kommen Familien und Organisationen aus ganz Europa und Israel im Rahmen des STXBP1-Awareness Month zu einer einzigartigen Spendenaktion zusammen. Jedes Land veranstaltet seine eigene Spendenaktion. Die meisten organisieren Staffelläufe der Hoffnung, um das Bewusstsein zu schärfen und bahnbrechende Forschungsarbeiten zu unterstützen.
Weitere Infos auf der Webseite von ESCO:
Webseite ESCO
STXBP1 Awareness Month 2024
September ist schon seit 2017 der STXBP1 Awareness Month.
In 2024 findet der STXBP1 Awareness Month vom 1. September bis zum "Giving Tuesday" am 3. Dezember 2024 statt.
Dieses Jahr wird die jährliche Veranstaltung neu gestaltet. Das Moto ist "Shine a Light on STXBP1".
Viele spannende Dinge sind geplant, wie z. B. die "Move to Cure"-Veranstaltung in den USA und Wanderungen weltweit, wie auch in Deutschland. Mitteilungen und Aktivitäten werden auf den Social-Media-Kanälen veröffentlicht. Bleiben Sie auf dem Laufenden und schauen Sie bei Facebook und Instagram nach.
Die gesammelten Spenden ermöglichen es, die erstaunliche Arbeit der STXBP1-Forscher fortzusetzen, um die Forschung nach besseren Behandlungen und einer möglichen Heilung für diese seltene genetische Krankheit voranzutreiben.
Die Veranstaltung "Move To Cure" gegen STXBP1-Störungen ist aus einer allerersten Spendenaktion hervorgegangen, einem 5-km-Lauf im Oktober 2017 in Ohio. Er wurde von Emmas Mutter Jennifer speziell für sie organisiert.
Infos unter: September Awareness Month 2024 in Amerika


Credit "Shine to light STXBP1": STXBP1 Foundation
Am 19. September 2024 trafen sich an die 70 Kinder und Jugendliche mit ihren Eltern an der Bike-Arena des Bergradler-Vereins in Oberthal.
Anlass war diesmal nicht nur das wöchentliche Radtraining, sondern auch der "STXBP1 Awareness Month September 2024". Der 1. Vorsitzender des Vereins erklärte den Kindern und Jugendlichen, dass es Menschen gibt, die an unheilbaren Krankheiten wie STXBP1-RD leiden und schilderte ihnen eindrucksvoll die Auswirkungen der Erkrankung. Zur Bekanntmachung der Erkrankung und zum Aufruf für Spenden für die STXBP1-RD Forschung zum Zwecke der Heilung bekamen die Kinder und Jugendlichen sodann Luftballons mit der Aufschrift STXBP1. Die Ballons präsentierten sie für ein Foto und durften sie nach dem Radtraining mit nach Hause nehmen. Ein herzliches Dankeschön an den Bergradler-oberthal.de Verein für diese tolle Aktion!








Am 28. September 2024 fand in Haidmühle eine Spendenveranstaltung statt. Trotz des unbeständigen Wetters wanderten 160 Teilnehmer 8 km durch die Haidmühler Wälder. Es war ein wunderschöner Tag, geprägt von vielen netten Gesprägen und strahlenden Gesichtern. Nach der Wanderung wurden alle mit leckerem Essen und erfrischenden Getränken versorgt. Durch die große Beteiligung konnten erneut auf STXBP1 aufmerksam gemacht und die Forschung unterstützt werden. Ein herzliches Dankeschön an alle, die diesen Tag besonders gemacht haben.


Über 30 Menschen machten sich in Hainert am 15. September 2024 zum Sponsorenlauf zur roten Quelle gemeinsam auf den Weg, um auf STXBP1 aufmerksam zu machen. Großer Dank gilt den Teilnehmern und den Sponsoren der Wanderung mit deren Hilfe der Verein STXBP1 e.V. und somit die Forschung unterstützt wird.


Am 7. September 2024 fand im Odenwald (Hessen) ein Charity Walk statt, um auf STXBP1-RD aufmerksam zu machen. An der Pfirsbacher Grillhütte verbrachten die Teilnehmer nach der Wanderung einen gemütlichen Tag mit vielen netten Gesprächen. Es war ein toller Tag im STXBP1 Awareness Month September.


STXBP1 Awareness Month 2023
Auch in 2023 widmeten wir den ganzen Monat September der STXBP1-RD (Rare Disorder).
Mit fortlaufenden Mitteilungen in Facebook und Instagram, und auch mit Aktivitäten an verschiedenen Orten in Deutschland, machten wir auf diese seltene STXBP1 Krankheit, an der 81 Kinder in Deutschland und 1109 weltweit leiden, aufmerksam.
Wir brachten STXBP1 Familien zusammen und sammelten Spenden mit Läufen und Fundraisern, um die STXBP1 Forschung zu unterstützen.
Familien riefen auf zum Mitmachen beim Sponsorenlauf oder Charity Wanderung in den Orten, wo ihre Familien wohnen.
Auch in Amerika war es das fünfte Jahr, dass der ganze Monat September der STXBP1-Enzephalopathie gewidmet wurde. Auch dort gab es wieder viele Aktivitäten.
Infos unter: September Awareness Month 2023 in Amerika





Credit STXBP1 e.V.
STXBP1 Awareness Month 2022
Der STXBP1 Awareness Monat war wieder im September.
Dies war das vierte Jahr, in dem in Amerika den ganzen Monat September der STXBP1-Enzephalopathie gewidmet wurde. Es gab wieder viele Aktivitäten.
Eltern, Großeltern, Freunde und Bekannten setzten sich ein mit dem "Move to Cure" ein, um das Bewusstsein für STXBP1-Störungen zu schärfen und Spenden für die STXBP1-Forschung zu sammeln und die Gemeinschaft der STXBP1 Familien zusammenzubringen.
Infos unter: September Awareness Month in Amerika
Auch in Deutschland fand zum ersten Mal ein Event im Monat September statt, um die STXBP1 Erkrankung, an der 71 Kinder in Deutschland leiden, bekannt zu machen.
Familien, Freunde und Bekannten wanderten oder radelten in ganz Deutschland, um auch für den STXBP1 Verein Spenden zu sammeln, damit die STXBP1 Forschung unterstützt werden kann.
Im Vereinsshop könnten für dieses Event im September T-Shirts für Kinder erworben werden.
Shop des Vereins: T-Shirts für den Awareness Month September in Deutschland



Credit STXBP1 e.V.
Familien riefen auf zum Mitmachen beim Sponsorenlauf oder Charity Wanderung in den Orten, wo ihre Familien wohnen.
Verschiedene Events wurden im Monat September auf Facebook und Instagramm bekannt gegebern.
STXBP1 Awareness Month 2021
Der STXBP1 Awareness Monat ist September, da sich das Gen STXBP1 auf Chromosom 9 befindet und der September der neunte Monat des Jahres ist.
Dies war das dritte Jahr, in dem der gesamte Monat September den STXBP1-assoziierten Störungen gewidmet wurde, mit vielen laufenden Kommunikationen und Aktivitäten um mehr Bewusstsein für diese seltene, genetisch bedingte Erkrankung zu schaffen.
Der STXBP1-Gipfel und Virtuelles Familientreffen fand vom 17. September bis 19. September 2021 statt!
Wissenschaftliche Updates, Workshops und gesellige Stunden! Themen sind u. a. klinische Präsentation, Therapieentwicklung, Bereitschaft für klinische Studien, Unterstützung von Betreuern, Interessenvertretung.
Hier finden Sie das Programm:
Programm STXBP1 Summit 2021
Research Update (auf Deutsch) mit Dr. Steffen Syrbe, Universitätsklinikum Heidelberg und Kim Thalwitzer Universitätsklinikum Heidelberg fand Sonntag, 19. September 2021 um 14.00 Uhr statt.
Das Video (auf Deutch) können Sie hier anschauen:
Video aufgenommen von STXBP1 Foundation - STXBP1 Summit 2021. Video hier publiziert mit Genehmigung von STXBP1 Foundation und Teilnehmern.
STXBP1 Awareness Month 2020
Der STXBP1 Awareness Monat ist September, da sich das Gen STXBP1 auf Chromosom 9 befindet und der September der neunte Monat des Jahres ist.
2019 schlug Jennifer Clatterbuck (STXBP1-Foundation) vor, einen STXBP1-Sensibilisierungsmonat zu starten. Es wurde sehr gut aufgenommen und ist jetzt zu einer jährlichen Veranstaltung geworden.
Dies ist das zweite Jahr, in dem der gesamte Monat September den STXBP1-assoziierten Störungen gewidmet wird, mit vielen laufenden Kommunikationen und Aktivitäten um mehr Bewusstsein für diese seltene, genetisch bedingte Erkrankung zu schaffen.
Eine jährliche Veranstaltung "Move to Cure" bringt die STXPB1 Gemeinschaft zusammen, dieses Jahr virtuell wegen Corona.
Die STXBP1-Stiftung ist eine Elternorganisation, die dem Fonds Geld zur Verfügung stellt, welches der Forschung zugutekommt. Die Organisation besteht aus einem vielfältigen Team von Familien und ihren Unterstützern, Wissenschaftlern und medizinischen Fachleuten und Forschern. Alle Spenden an STXBP1 Foundation kommen der Forschung zugute. Die Forscher sind bestrebt, ein Heilmittel für die sogenannte "STXBP1 Epileptic Encephalopathy" zu finden (siehe auch button "Breaking News").
Durch die Förderung von Partnerschaften mit Ärzten, Forschern und anderen Stiftungen tauschen sie Erfahrungen und Erkenntnisse aus, um das Bewusstsein für diese seltene, genetisch bedingte Erkrankung zu schärfen und die Zeit bis zur Heilung zu verkürzen. Sie glauben, dass sie durch ihre Arbeit den Weg zu verbesserten Therapien und letztlich zur Beendigung der STXBP1-Enzephalopathie finden werden.
Die internationale STXBP1-Familie ist enorm auf über etwa 1100 Familien angewachsen, die vereint und stark sind, und ihre Zahl wird nur noch weiterwachsen, wenn andere Familien auch Zugang zu Gentests haben.
Webinare im Monat September 2020
Die momentane Forschung an STXBP1 wurde in Webinaren im September 2020 online vorgestellt.
1. Designing an STXBP1 Natural History Study and Verhage Lab Updates
Matthijs Verhage und Mitglieder seines Labors haben das Design einer neuen STXBP1-Naturkundestudie diskutiert und zusätzliche Updates aus dem Labor geliefert. Matthijs Verhage, PhD, ist ordentlicher Professor und Leiter der Abteilung für Funktionelle Genomik am VU University Medical Center in Amsterdam. Er ist Mitbegründer und stellvertretender Vorsitzender des niederländischen NeuroBsik Mouse Phenomics-Konsortiums, Partner des europäischen Forschungskonsortiums EU-Synapse und Gründer und stellvertretender Vorsitzender des kürzlich ausgezeichneten H2020-Konsortiums COSYN. Simon Houtman, Doktorand sprach über die Verwendung des EEG für bessere Behandlungsentscheidungen beim STXBP1-Syndrom. Annemiek van Berkel, Doktorandin & Hanna Lammertse, Doktorandin berichteten über die Folgen von STXBP1-Mutationen auf zellulärer Ebene.
Das Video können Sie hier anschauen: Video Matthijs Verhage, Simon Houtman, Annemiek van Berkel und Hanna Lammertse, Amsterdamn
Die deutsche Sprachdatei (pdf) können Sie hier downloaden: Deutsche Übersetzung
2. ASO and Genetic Therapies as a Therapeutic Approach for STXBP1 Encephalopathy
Ben Prosser gab einen Überblick über genetische Therapien als einen möglichen Behandlungsansatz für STXBP1. Er ging auch auf seine aktuelle Forschung über ASOs ein. Ganna Balagura, MD von der Universität Genua, diskutierte SINEUPs als neuartigen gentherapeutischen Ansatz für STXBP1. Ben Prosser, PhD, ist Assistenzprofessor für Physiologie an der University of Pennsylvania Perelman School of Medicine. Dr. Prosser weitete seine Forschung über Herzerkrankungen auch auf die Untersuchung von Störungen der Neuroentwicklung aus, als er Elternteil einer Tochter mit STXBP1-Enzephalopathie wurde.
Das Video können Sie hier anschauen: Video Ben Prosser und Ganna Balagura
Die deutsche Sprachdatei (pdf) können Sie hier downloaden: Deutsche Übersetzung
3. STXBP1 Update from Weill Cornell
Jacqueline Burré, PhD, Debra Abramov und Noah Guiberson präsentierten einen 282 STXBP1-Patientenbericht und diskutierten auch Small Molecule Therapy Approaches.
Dr. Zach Grinspan berichtete über den aktuellen Stand des Pilotversuchs für eine potenzielle niedermolekulare Behandlung, 4-Phenylbutyrat, dass in Burrés Labor identifiziert wurde (siehe auch button "Breaking News").
Das Video können Sie hier anschauen: Video Jacqueline Burré, Debra Abramow, Noah Guiberson und Zach Grinspan
Die deutsche Sprachdatei (pdf) können Sie hier downloaden: Deutsche Übersetzung
4. How Different Neurons in the Brain Contribute to STXBP1 Encephalopathy
Mingshan Xue, PhD, ist Assistenzprofessor in der Abteilung für Neurowissenschaften und am Jan und Dan-Duncan-Institut für neurologische Forschung am Baylor College of Medicine. Dr. Xue diskutierte aktuelle Studien über den Beitrag verschiedener Arten von Neuronen zur STXBP1-Enzephalopathie.
Das Video können Sie hier anschauen: Video Mingshan Xue
Die deutsche Sprachdatei (pdf) können Sie hier downloaden: Deutsche Übersetzung
5. What we know about STXBP1: An Update
Ingo Helbig, MD des Kinderkrankenhauses von Philadelphia, informierte über über die Biologie und Funktion von STXBP1, sowie über die klinische Präsentation.
Sarah McKeown, MS, LCGC, gab einen Überblick über die ENGIN-Klinik (Multidisziplinäre Epilepsie-Neurogenetik-Initiative) und ihre Arbeit mit STXBP1-Enzephalopathie-Patienten.
Das Video können Sie hier anschauen: Video Ingo Helbig und Sarah McKeown
Die deutsche Sprachdatei (pdf) können Sie hier downloaden: Deutsche Übersetzung
6. Simons Searchlight and STXBP1 Registry Update
Wendy Chung, PhD, Direktorin für klinische Forschung bei der Simons Foundation und Professorin für Pädiatrie an der Columbia University, gab ein informatives Update zu Simons Searchlight und STXBP1.
Das Video können Sie hier anschauen: Video Wendy Chung
Die deutsche Sprachdatei (pdf) können Sie hier downloaden: Deutsche Übersetzung
459798354
Spendenaktion im Awareness Month 2020
Im September 2020 wurden auf Facebook mehrere Spendenaktionen für STXBP1 Disorders gestartet. Dies war eine der vielen Aktivitäten im STXBP1 Awareness Month.
Es kamen 65,000 $ zusammen, was fast einer Verdreifachung gegenüber dem letzten Jahr entspricht. Es nahmen mehr als 250+ Menschen in über 30+ Staaten und 4 verschiedenen Ländern teil. Diese Beiträge tragen dazu bei, den Kampf um eine Heilung von STXBP1 zu verbessern
Wollen Sie sich dieser Aktion noch anschließen und einen guten Zweck unterstützen? Ihr Beitrag wird Wirkung zeigen, egal, was Sie spenden. Jedes kleine bisschen hilft. STXBP1 Foundation dankt Ihnen für Ihre Unterstützung.
Klicken Sie hier: Spenden.
Die STXBP1 Farbe ist kastanienbraun
Warum kastanienbraun?
Kastanienbraun, weil Lila für Epilepsie und Blau für Autismus und Grün für Zerebralparese steht. Bei STXBP1-Kindern wird meistens Epilepsie diagnostiziert, und einige haben auch Autismus, und auch einige Zerebralparese.
Warum Lila (purple) für Epilepsie?
Das Konzept des "Purple Day" wurde von einer 9-jährigen namens Cassidy Megan initiiert und war von ihrem eigenen Kampf gegen Epilepsie motiviert. Die Epilepsie-Vereinigung von "Nova Scotia" half bei der Entwicklung von Cassidys Idee. Die erste Veranstaltung zum "Purple Day" fand am 26. März 2008 statt und ist jetzt als Kampagne zum "Purple Day" für Epilepsie bekannt.
Warum Blau (blue) für Autismus?
Der 2. April ist der Welttag des Bewusstseins für Autismus, und viele Menschen tragen die Farbe Blau, um auf die Entwicklungsstörung aufmerksam zu machen.
Warum Grün (green) für Zerebralparese?
Zu Ehren des "Cerebral Palsy Awareness Month" tragen viele Menschen Grün. Warum Grün? Die Farbe wurde ausgewählt, um Jugendlichkeit und neues Wachstum, sowie die Hoffnung auf Fortschritte bei der Behandlung der Krankheit und Akzeptanz wiederzuspiegeln.
Das Gen STXBP1 liegt auf Chromosom 9
STXBP1 ist ein Gen, das an der Entwicklung des Gehirns und der Neurotransmitter-Signalübertragung beteiligt ist. Es ist auch als Munc18-1 bekannt. Veränderungen im STXBP1-Gen (Mutationen) können zu Epilepsie, geistiger Behinderung, Entwicklungsverzögerung, Bewegungsstörungen und anderen Schwierigkeiten führen.
Bei einem Kind mit Merkmalen von STXBP1-assoziierten Störungen, wie z.B. früh einsetzender Epilepsie, kann ein Arzt einen Genpanel- oder Microarray-Test anordnen, der Mutationen oder Varianten im STXBP1-Gen identifiziert.
STXBP1-Varianten können auch durch einen neueren Gentest, namens Sequenzierung des gesamten Exoms, diagnostiziert werden (WES=Whole Exome Sequencing).
Die Patienten haben ein breites Spektrum an Symptomen. Die meisten Kinder haben ein gewisses Maß an kognitiven Beeinträchtigungen, die von leicht bis schwerwiegend reichen. Bewegungsstörungen und autistische Merkmale sind häufig. Viele Kinder haben Probleme mit dem Essen und Schlucken. Die meisten Patienten sind non-verbal, obwohl einige sprechen und⁄oder Zeichen geben.
STXBP1 Kinder entwickeln sich sehr unterschiedlich
Das Gen STXBP1 produziert ein wichtiges Protein (syntaxin-1) für die Neurotransmitter-Freisetzung.
Die Proteinfaltung ist aufgrund von stxbp1-Mutationen in der Aminosäuresequenz nicht korrekt möglich. Das produzierte Protein ist dadurch fehlerhaft oder nicht funktionsfähig.
Das Gen STXBP1 hat 20 Exons (diese produzieren das Protein) und STXBP1 Kinder haben Mutationen auf verschiedenen Loci des Gens STXBP1 (Orten, Stellen des Gens). Einige z.B. in Exon 2, andere z.B. in Exon 18, usw.
Dazu gibt es noch verschiedene Arten von Mutationen, wie Missense, Nonsense, Insertion, Deletion, Frameshift, Splicing. Eine gute Erklärung (auf Englisch) hierzu finden Sie hier; die Bilder zeigen, wie unterschiedlich diese Mutationen sind. Klicken Sie hier: Mutationen
Eine ganze Gendeletion klingt irgendwie beängstigend und klingt, dass sie schwerer sein muss als andere Mutationen. Alle Mutationen haben die gleiche Art von Effekt. Und der Effekt am Ende ist, dass es nur eine Kopie von STX gibt, die voll funktionsfähig ist. In neueren Studien wurde festgestellt, dass die meisten Mutationen Missense Mutationen sind, etwa 47 %. Kinder mit einer Missense Mutation scheinen einen milderen Verlauf zu haben, weil bei dieser Mutation das Protein noch bedingt funktionsfähig ist. Bei den anderen Mutationen ist das Protein unbrauchbar.
Kinder können die gleiche Mutation haben und sich dennoch unterschiedlick entwickeln.
Bei den Kindern, die Epilepsie haben, spielt sich die Epilepsie in verschiedenen Bereichen des Gehirns ab. Dadurch sind Bereiche im Gehirn gestört, Bereiche die verschiedene Aufgabe im Gehirn haben, z.B. Motorik, Emotionen, Gedächtnis, Sprache, um nur einige zu nennen. Das erklärt auch, warum ein Kind etwas Gelerntes wieder nicht mehr kann, wenn die Epilepsie wiederkommt. Wenn man die Art der Anfälle ansieht, hat STXBP1 tatsächlich etwas sehr Interessantes und Einzigartiges. Und das wirklich Einzigartige an STXBP1 ist, dass die epileptischen Anfälle bei den STX-Patienten unabhängig von ihrem Entwicklungsprofil zu sein scheinen. Und sie scheinen auch unabhängig davon zu sein, welche anderen Anfälle STX-Patienten vorher oder nachher gehabt haben.
STXBP1 Kinder sind non-verbal. Was versteht ein STXBP1 Kind?
Es gibt mehrere Bereiche des Gehirns, die eine entscheidende Rolle in Sprachproduktion und Sprachverständnis spielen.
Der Brocas Bereich ist eine Region im Frontallappen der dominanten Hemisphäre, normalerweise der linken Hemisphäre des Gehirns. Brocas Bereich ist mit Sprachproduktion und Artikulation verbunden.
Der Wernickes Bereich befindet sich im hinteren oberen Temporallappen der linken Gehirnhälfte. Wernickes Bereich enthält Motorneuronen, die am Sprachverständnis beteiligt sind.
Wernickes Gebiet ist ein kritisches Sprachgebiet, das über einen Nervenweg mit Brocas Gebiet verbunden ist, und am Verständnis der geschriebenen und gesprochenen Sprache beteiligt ist.
Das Sprachverständnis spielt sich also vor allem im Wernickes Bereich des Gehirns ab, obwohl es viel komplexer ist und noch andere Bereiche eine wichtige Rolle spielen.
Viele Kinder mit Sprachverzögerungen haben orale motorische Probleme. Hörprobleme können auch die Sprache beeinträchtigen.
Was ein STXBP1 Kind versteht ist oft schwer zu beurteilen, und es ist nur durch genaue Beobachtung des einzelnen Kindes zu beurteilen.
Jährlich wird am 28. Februar das Bewusstsein für mehr als 8000 seltene Erkrankungen geschärft.
Weltweit sind über 300 Millionen Menschen betroffen. Diese Kampagne von:
Rare Disease Day,
NORD und
EURORDIS
beleuchten seltene Krankheiten und setzen sich für
eine gesundheitliche Chancengleichheit für alle Menschen mit einer seltenen Krankheit ein.
In 2025 ist der Rare Diseases Day wieder am 28. Februar.
Im Jahr 2025 hat sich einiges geändert: 33 Selbsthilfegruppen sind nun Teil des RDR25, organisiert von der Syngap1 Elterngruppe e.V..
Auch in Oberthal wird wieder ein RDR in 2025 organisiert. Weiter Infos demnächst hier, und auf Facebook und Instagram.
Video Copyright Syngap1 Elterngruppe e.V.

Credit Gilberte Schnur
Der Rare Diseases Run (RDR) ist der größte Online-Lauf für seltene Erkrankungen in Europa! Zum 4. Mal fand diese tolle Veranstaltung im Zeichen der Inklusion statt.
Die Elternhilfe Syngap e.V als Initiator und weitere 33 Vereine stehen unter "Laufen macht glücklich" hinter diesem Projekt und möchten damit die Aufmerksamkeit auf Menschen mit seltenen Erkrankungen richten.
Seltene Erkrankungen sind vielfältig und nicht immer sichtbar. Seltene Erkrankungen sind nicht so selten, wie viele denken: 400 Millionen Menschen auf der Welt leben mit einer der mehr als 7.000 seltenen Erkrankungen. Das sind etwa 5 % der gesamten Weltbevölkerung. Davon leben ca. 4 Millionen Menschen in Deutschland. Mit diesem Lauf haben 33 Vereine den Focus auf die Waisen der Medizin mit ihren besonderen Herausforderungen gerichtet.
Mehrere Läufe wurden von der STXBP1 Gemeinschaft organisiert.


Credit Gilberte Schnur


Credit STXBP1 e.V.


Credit STXBP1 e.V.
Ein ganz besonderer Tag der seltenen Krankheiten in 2024: der 29. Februar ist der Rare Diseases Day in 2024.
Der Tag der Seltenen Krankheiten im Jahr 2024 fand am 29. Februar statt.
Der STXBP1 e.V. gehörte im Jahr 2024 zu den ausgewählten gemeinnützigen Vereinen. STXBP1 Familien liefen am "Rare Diseases Day", um Spenden zu sammeln, die an 22 gemeinnützigen Organisationen verteilt wurden.
Die Anmeldung war schon ab Oktober 2023 möglich! Es gab viele Möglichkeiten für Erwachsene und Kinder, für Schulen und Firmen, um an diesem Ereignis teilzunehmen. Anmeldung war hier:
Mehrere Pakete standen zur Auswahl!


Credit Syngap Elternhilfe e.V. ‐ Credit Gilberte Schnur


Credit Gilberte Schnur
Was war nun los vom 26. Februar bis zum 3. März 2024?


Credit CDU Oberthal
Es ist der 2. März 2024.
Die Kirchenglocken am alten Bahnübergang in Oberthal läuten 10 Uhr und verkünden nicht nur die Stunde, sondern auch den Startschuss für den "Rare Diseases Run 2024", organisiert von "Syngap1 Elternhilfe e.V." und "Laufen Macht Glücklich", um auf seltene Erkrankungen aufmerksam zu machen und Unterstützung für Betroffene zu mobilisieren.
Die Einnahmen des "Rare Diseases Run 2024" kommen insgesamt 22 gemeinnützigen Vereinen, worunter auch STXBP1 e.V., zugute.
Ein besonderes Event in Oberthal, organisiert von der politischen Partei "CDU Oberthal" und Gilberte Schnur, Mitglied des STXBP1 Vereins und Oma vom STXBP1 Kind Jannek, bringt die Gemeinde zusammen, um auf die seltene STXBP1 Krankheit aufmerksam zu machen. Zahlreiche Berichterstattungen erscheinen in Zeitungen zum Event in Oberthal, im Saarland, dem kleinsten Bundesland Deutschlands, in dem 2 Kinder mit der Diagnose STXBP1 betroffen sind; Zwei von insgesamt 81 Kindern in Deutschland.
Bei gutem Wetter sammeln sich Teilnehmerinnen und Teilnehmer am alten Bahnübergang, wo sie von Luftballons mit dem Symbol STXBP1 begrüßt werden. Während sich immer mehr Läufer aufwärmen und eintreffen entstehen zahlreiche Foto-Momente, die die Solidarität und Gemeinschaft unter den Anwesenden festhalten.
Der Lauf erstreckt sich über vier Stunden – von 10 bis 14 Uhr – und bietet sowohl ambitionierten Läufern als auch gemütlichen Spaziergängern die Möglichkeit, sich für eine gute Sache einzusetzen. Nachdem die Läufer das Ziel erreicht haben, wartet auf alle Teilnehmenden eine wohlverdiente Stärkung mit Würstchen und Getränken.
Namhafte Unterstützer dieses Laufs sind die Globus Stiftung, Lotto Rheinland-Pfalz, die Oskar Killinger Stiftung und natürlich die CDU Oberthal, die alle Kosten der Startgelder und Verpflegung übernimmt. Ihr Engagement ermöglicht es, ein Zeichen der Hoffnung zu setzen und gleichzeitig wichtige Mittel für Forschung und Unterstützung für die seltene Erkrankung STXBP1 und andere seltene Erkrankungen zu sammeln.
In Oberthal schlägt das Herz der CDU für seltene Erkrankungen, Gemeinschaft, Inklusion und Solidarität.


Credit Gilberte Schnur
Nicht nur in Oberthal wurde gelaufen!
Zu Pferd wurden Strecke zurückgelegt. Familien trafen sich zum Lauf in Höchst im Odenwald und liefen In Hainert.
Das Mainathlon Team Eltmann lief für STXBP1 e.V.


Credit STXBP1 e.V.
Tag der seltenen Erkrankungen am Universitätsklinikum Homburg, Saarland
Zwei Vereine des RDR24 waren anwesend: STXBP1 und MFSH (Morbus Fabry).

Credit STXBP1 e.V.
Rare Diseases Run fand in 2023 vom 28. Februar bis zum 5. März 2023 statt.
Der Verein STXBP1 e.V. unterstützte diesen Lauf.
Rare Disease Run 2023

Credit Gilberte Schnur
Rare Disease in Amerika, STXBP1 Foundation:
Rare Disease Day der STXBP1 Foundation in Amerika
13. Breaking News: Gentherapie mit Capsid-002 / Pilot Project mit 4-Phenylbutyrat |
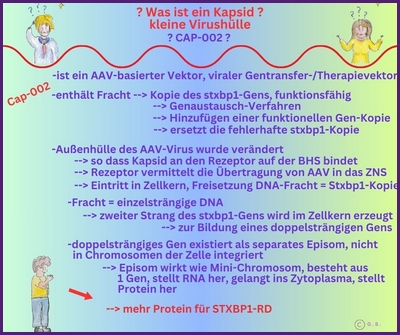
Gentherapie mit Capsid-002 von Capsida Biotherapeutics
Capsida
Biotherapeutics, ein Biotech-Unternehmen, hat bekannt gegeben, dass die US Food and Drug Administration
Arzneimittelbehörde (FDA) einen Orphan-Drug-Status für eine potenzielle Gentherapie für STXBP1-bezogene Erkrankungen erteilt hat.
Capsida führte diese Studien in Zusammenarbeit mit Mingshan Xue, Ph.D., außerordentlicher Professor für Neurowissenschaften und Molekular- und Humangenetik am Baylor College of Medicine, sowie Mitglied der Cain Foundation Laboratories und des Duncan Neurological Research Institute am Texas Children's Hospital durch. Eine klinische Studie mit Capsid-002 ist im dritten Quartal 2025 geplant. Die Rekrutierung hat in Philadelphia im Juli 2025 begonnen.
Alle Infos und Updates zu der klinischen Studie finden Sie hier:
Erste Gene Therapy für STXBP1-RD
Weitere Infos finden Sie auf folgenden Webseiten:
Orphan-Drug-Status Capsida
Pipeline for Rare and Common Diseases Capsida
Press Releases Capsida
Capsida Publications
Auch auf der STXBP1 Foundation Webseite gibt es ebenfalls die neuensten Informationen zu der klinischen Studie:
Oral presentation Capsida at ASGCT 2025
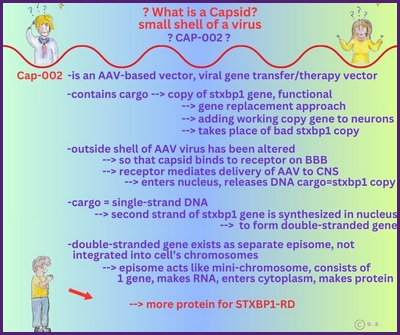
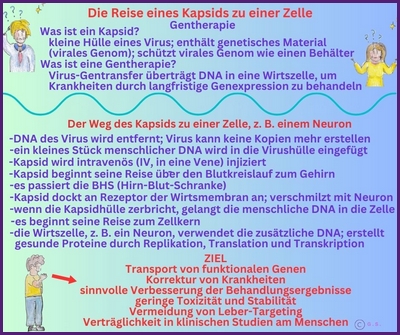
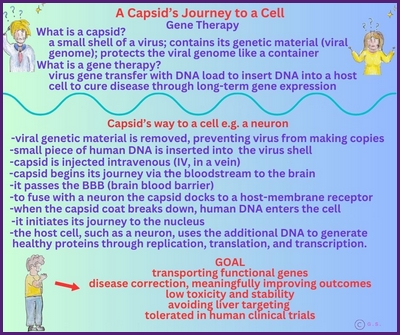
Copyright Gilberte Schnur
Preprint zu der Glycerinphenylbutyrat (Ravicti®) klinischen Studie wurde veröffentlicht.
Ravicti® hat weltweit den ersten Orphan-Status für ein Medikament gegen STXBP1 bekommen. Das Egebnis der klinischen Studie liegt in Preprint vor. Weitere Infos auf:
Preprint klinische Studie für Ravicti®
Immedica hat Glycerinphenylbutyrat (Ravicti®) bei STXBP1-bedingten Erkrankungen den Orphan-Status erteilt
Dies ist weltweit der erste Orphan-Status für ein Medikament gegen STXBP1!
Weitere Infos auf:
Orphan Drug Status für Ravicti®
Link zu Immedica (Vertrieb von Ravicti®):
Immedica gibt Orphan Drug Status für Ravicti® bekannt
Update Ergebnisse der 4-Phenylbutyrat Studie in New York
Forscher, u.a. Dr. Zachary Grinspan, vom Weill Cornell Medicine, zeigten auf dem 1. Europäischen Gipfel in Milam am 17. Mai 2023 weitere positive Ergebnisse der Ravicti Studie.
Sie können den Vortrag auf der Webseite der STXBP1 Foundation ansehen: EU Summit 2023 in Milan
Vidoes STXBP1 EU Summit 2023
Die deutsche Übersetzung (pdf) der Präsentation von Zach Grinspan über Ravicti auf der 1. Europäischen Gipfel in Milan am 17. Mai 2023 können
Sie hier downloaden: Deutsche Übersetzung des Videos Ravicti
(Credit Daten und Grafik: STXBP1disorders.org)
Erste Ergebnisse der 4-Phenylbutyrat Studie
Forscher, u.a. Dr. Zachary Grinspan, vom Weill Cornell Medicine, haben die ersten positiven Ergebnisse der Ravicti Studie gesammelt.
Die Ergebnisse wurden am 19. - 20. August 2022 auf der STXBP1 Summit+ in Philadelphia bekannt gegeben.
Sie können seinen Vortrag auf der Webseite der STXBP1 Foundation ansehen: Research Roundtable - Clinical Update
Vidoes STXBP1 Summit+ 2022
Hier mehr Info über die Studie (auf Englisch): Klinische Studie mit Glycerol Phenybutyrat.
Neue Therapie Studie für STXBP1 in US mit 4-phenylbutyrat
Forscher von Weill Cornell Medicine leiten die Entwicklung eines Pilot-Behandlungsprotokolls für STXBP1-Enzephalopathie mit Epilepsie mit einem finanziellen Zuschuss vom "The Orphan Disease Center" und "Clara Inspired".
In einer revolutionären Studie fanden Dr. Jacqueline Burré und ihre Kollegen heraus, dass ein Medikament namens Phenylbutyrat als chemisches Chaperon wirken kann, um das funktionelle STXBP1 zu stabilisieren. Diese Studie zielt darauf ab, Phenylbutyrat als Behandlungsoption in die klinische Praxis einzuführen.
Unter der Leitung von Dr. Zachary Grinspan, Direktor der Pädiatrischen Epilepsie, werden die Forscher von Weill Cornell Medicine zehn Kindern mit STXBP1-Enzephalopathie Phenylbutyrat verabreichen. Es werden auch Interviews mit Familienmitgliedern durchgeführt, um mehr über ihre Erfahrungen mit STXBP1 und mit dem Studienmedikament zu erfahren. Die Forscher haben drei Ziele: die Erprobung eines Evaluierungsprotokolls, die Bewertung des Wirksamkeitspotenzials und die Beurteilung der Serumspiegel aktiver Metaboliten in der Patientenpopulation.
Das Medikament wirkt im Labor, aber die Forscher wissen noch nicht, ob es bei den Patienten wirkt. Bei dieser First-in-Disease-Studie geht es wirklich um Sicherheit und Verträglichkeit, aber die Forscher werden auch prüfen, ob es einen klinischen Nutzen gibt.
Hier mehr Info (auf Englisch): Forschung mit Phenybutyrat.
Wie wirkt 4-phenylbutyrat?
Phenylbutyrat ist ein Medikament, das als chemisches Chaperon wirken kann, um das Protein STXBP1 bei der Proteinfaltung zu stabilisieren.
Eine Proteinfaltung ist aufgrund von stxbp1-Mutationen in der Aminosäuresequenz bei STXBP1 Kindern nicht möglich. Proteinfaltung ist der Prozess, durch den eine Proteinstruktur ihre funktionelle Form oder Konformation annimmt.
Durch die Faltung in eine spezifische 3-dimensionale Form ist das Protein Syntaxin-1 in der Lage, seine biologische Funktion zu erfüllen, was notwendig ist, damit die Neuronen im Gehirn miteinander kommunizieren und richtig funktionieren können.
Syntaxin-1 ist Teil des SNARE-Komplexes, der die Vesikelfusion in die Neuronenzellmembran vorbereitet und die Freisetzung des Neurotransmitters entlang der Synapsen erleichtert. Eine Änderung der Neurotransmitterspiegel kann zu einer unkontrollierten Aktivierung (Erregung) von Neuronen im Gehirn führen, was zu epileptischen Anfällen und andere Störungen führt.
Syntaxin-1 ist für die Transmitterfreisetzung von der Präsynapse eines Neurons zur Postsynapse eines anderen Neurons erforderlich. (Es ist der Schlüsselmechanismus, mit dem Neuronen miteinander kommunizieren.).
(siehe auch die Bilder auf der stxbp1.de Website, button "Was ist STXBP1?")
Was ist 4-Phenylbutyrat?
4-Phenylbutyrat ist ein chemisches Chaperon, das dazu in der Lage ist:
- Proteinspiegel von Munc18-1 (stxbp1) zu stabilisieren und neuronale Defizite zu korrigieren
- die Neurotransmitter-Freisetzung zwischen den Synapsen der Neuronen zu erhöhen bzw. zu verbessern
- unsachgemäß (falsch) gefaltete Proteine zu stabilisieren bzw. zu korrigieren
- betroffene Proteine effizienter zu falten und sie zum geeigneten intrazellulären oder extrazellulären Ziel zu transportieren
Was sind Chaperone? Chaperone sind Proteine, die die neu synthetisierten Proteine bei der Faltung bzw. die neu synthetisierten Proteine "helfen", sich korrekt zu falten.
Eine erfolgreiche 4-Phenylbutyrat-Therapie könnte daher STXBP1 Kindern helfen, da ihnen das Protein Syntaxin-1 fehlt oder es fehlerhaft ist.
Das 4-Phenylbyturat könnte die Defizite bei der Neurotransmitterfreisetzung mit Munc18-1 (stxbp1) Mutationen signifikant beheben und die synaptische Funktion bei den STXBP1 Kindern wieder auf ein normales Funktionsniveau zurückbringen.
Hoffen wir auf eine Heilung für die STXBP1 Kinder. Phenylbutyrat wird nun an 10 STXBP1 Kinder getestet.
14. Aktuelle Forschungen :: "Ermutigt von der Forschung ‐ Vereint durch die Hoffnung" |
Capsida Biotherapeutics
Neuigkeiten auf der Jahrestagung der American Society of Gene & Cell Therapy (ASGCT) im Mai 2024
Capsida hat zwei neue Programme: Parkinson-Krankheit im Zusammenhang mit GBA-Mutationen (PD-GBA) und neue Daten aus Capsidas hundertprozentigen Programmen zu PD-GBA und genetischer Epilepsie aufgrund von STXBP1-Mutationen. Beide Programme ermöglichen die Unterstützung der Einleitung klinischer Studien im ersten Halbjahr 2025. Die AAV-Gentherapie korrigiert neurologische Phänotypen mit klinisch relevanten Dosen in einem Mausmodell der STXBP1-bedingten Entwicklungs- und epileptischen Enzephalopathie.
Weitere Info:
Neue Daten zu Gentherapieprogrammen bei genetischer Epilepsie und Parkinson-Krankheit auf der Jahrestagung ASGCT 2024
Weitere Infos unter 13. Breaking News
Encoded Therapeutics
Fortschritte präklinischer Programme zur Einbeziehung von STXBP1-bedingten Erkrankungen (STXBP1-RD)
Mehrere Pipeline-Programme: Fortschritte präklinischer Programme für das Angelman-Syndrom, das Lennox-Gastaut-Syndrom (LGS), STXBP1-bedingte Erkrankungen (STXBP1-RD), die Alzheimer-Krankheit (MAPT) und neuropathische Schmerzen (SCN9A) sowie Innovationen in der Vektor-Engineering-Plattform des Unternehmens.
Weitere Info:
Fortschritte präklinischer Programme zur Einbeziehung von STXBP1-RD auf der Jahrestagung ASGCT 2024
Ganganalyse von Personen mit STXBP1-RD
Studie zu motorischen Problemen, einschließlich Gangstörungen
Die Studie wird am Universitätsklinikum in Antwerpen, Belgien, von Prof. Sarah Weckhuysen und Dr. Hannah Stamberger, zusammen mit Prof. Ann Hallemans von der Abteilung für Physiotherapie durchgeführt.
Das Ziel ist es, die Art und Entwicklung von Ganganomalien und funktioneller Mobilität bei Patienten mit STXBP1-RD herauszufinden. Das Ergebnis der Studie kann für die klinische Nachsorge und für eine mögliche Zukunft nützlich sein.
Zwei Studienbesuche in Antwerpen sind erfolderlich für eime Teilnahme an der Studie. Es wird eine neurologische Untersuchung und eine im Ganglabor 3D-Ganganalyse durchgeführt. Zwei Fragebögen müssen von den Eltern ausgefüllt werden.
Das Nonsense-Mausmodell
Neu in der STXBP1-Forschung: Das Nonsense-Mausmodell
Die STXBP1-Stiftung arbeitet mit Jackson Labs zusammen, um ein Nonsense-Mausmodell zu entwickeln. Um Erkenntnisse über genetische Störungen zu gewinnen, sind Forscher auf präzise Werkzeuge angewiesen, die sowohl Tiermodelle als auch In-vitro-Modelle umfassen. Bis vor kurzem fehlte ein Mausmodell, das Nonsense-Mutationen reproduziert. Nonsense-Mutationen treten in etwa 23 % der Fälle auf und verursachen Probleme, weil sie die Bildung wichtiger Proteine zu früh stoppen. Dieses Nonsense-Mausmodell wird die Auswirkungen von Nonsense-Mutationen nachahmen und wertvolle Erkenntnisse über die Krankheit und mögliche Behandlungsmöglichkeiten liefern.
Die Schaffung eines Nonsense-Mausmodells für STXBP1 bietet neue Hoffnung für Menschen und Familien, die von STXBP1-Nonsense-Mutationen betroffen sind.
Hier mehr Info zum Nonsense-Mausmodell : Nonsense-Mausmodell mit Jackson Labs
BRAINMODEL Projekt
BRAINMODEL Projekt mit Professor Verhage und weiteren Teams, Amsterdam:
Ein neues Konsortium bestehend aus Forschern von sechs niederländischen Wissensinstitutionen entwickeln neue Forschungsmethoden zur Verbesserung der Behandlung neurologischer Entwicklungsstörungen.
Neue stammzellbasierte Methoden werden eingesetzt, um Diagnostik und Therapieentscheidungen zu verbessern und neue Therapien zu entwickeln.
Die BRAINMODEL Forschung basiert auf patienteneigenen Zellen. Diese werden in einer Kulturschale zu lebende Nervenzellen, die den neuralen Netzwerken im menschlichen Gehirn sehr ähnlich sind, umgeändert.
Dies wird als "pluripotente Stammzelltechnologie (iPSC)" bezeichnet. Diese Technologie ermöglicht menschliche Krankheiten besser zu verstehen und für diese personalisierte Behandlungen zu finden.
IPSC-basierte Strategien sind besonders vielversprechend für Entwicklungsstörungen des Gehirns.
Acht Kinder wurden jetzt in diesem Projekt aufgenommen. Die Untersuchungen beinhalten EEGs, Bluttests und x-rays der Hände, um die Knochendichte zu messen und den Mineralgehalt in den Knochen, wie z.B. Kalzium, zu messen. Wenn mit der Nahrung nicht genügend Kalzium aufgenommen wird, entzieht der Körper es den Knochen, um eine normale Zellfunktion zu gewährleisten, was zu Osteoporose - was bei STXBP1 Kindern vorkommt - führen kann.
Hier mehr Info zum BRAINMODEL Projekt : Webseite BRAINMODEL
Forscher versuchen den Verlust des STXBP1 Proteins, verursacht durch eine STXBP1 Gen Mutation, zu korrigieren. Sie forschen auf der Ebene der DNA, der RNA oder der Protein Ebene. Hier mehr Info (auf Englisch): Aktuelle Forschungen.
Baylor College of Medicine / Texas Children's Hospital
Das Labor von Dr. Mingshan Xue untersucht das Verhältnis zwischen synaptischer Erregung und Hemmung im Gehirn von Mausmodellen mit STXBP1-Enzephalopathie in der Großhirnrinde. Es wird versucht herauszufinden, warum ein Mangel an STXBP1 Protein zu Krampfanfällen, Verhaltensstörungen und kognitiven Defiziten führt.
Nemametrix / IN Vivo Biosysteme
Chris Hopkins forscht an humanisierten Würmern und Zebrafischen, um die Pathogenität bestimmter genetischer Varianten und die Beziehung zum Phänotyp, wie zum Beispiel von stxbp1, zu untersuchen.
VU organisierte Klinik-Tage
Das "Department of Functional Genomics" am "Centre for Neurogenomics and Cognitive Research" an der "Vrije Universiteit" und am "Amsterdam University Medical Centre" führt "Clinic Days" durch. STXBP1 Patienten und deren Familien können sich gegenseitig kennenlernen und mehr über STXBP1 und die Forschung der VU erfahren. Es werden detaillierte klinische Profile und EEGs der Patienten erstellt. Ziel ist es gemeinsame und nicht‐gemeinsame Symptome der STXBP1 Patienten zu erstellen. Diese Erkenntnisse werden mit Studien an Tier− und Zellmodellen für STXBP1-Enzephalopathie verknüpft. Die VU arbeitet mit "Filadelfia", dem Dänischen Epilepsiezentrum, und mit dem "Sørensen‐Labor" an der Universität in Kopenhagen zusammen.
Das Labor von Dr. Verhage hat Modellsysteme für Mäuse und menschliche Neuronen entwickelt. Sie untersuchen das Ungleichgewicht von Erregung und Hemmung bei der STXBP1-Störungen. Um spezifische Patientenphänotypen und genetische Patientenprofile zu untersuchen verwenden sie induzierte pluripotente Stammzellen, iPSC-Zellen. IPSCs werden aus Haut oder Blut gewonnen, die von STXBP1 Patienten und Familienmitgliedern gespendet werden.
Universität von Genova & INSTITUTO G. GASLINI
Dr. Federico Zara und Dr. Pasquale Striano am "IRCCS Istituto Giannina Gaslini" und der Universität Genua erforschen einen genetischen Therapieansatz namens SINEUPs für STXBP1-Störungen. Ganna Balagura aus Genua untersucht ebenfalls zusammen mit Professor Verhage (VU Amsterdam) SINEUPs an menschlichen Gehirnzellen. STXBP1-Sineups erhöhen die stxbp1-Proteinkonzentration um 50 % sowohl in gesunden Nicht-Hirnzellen als auch in gesunden menschlichen Neuronen.
Universität Oslo Zentrum für Molekulare Medizin Norwegen
Dr. Esquerra - University of Oslo - und ihre Gruppe verwenden den Zebrafisch als Modell, um die Gehirnfunktion zu untersuchen und epileptische Anfälle zu verstehen. Das Esquerra‐Labor versucht, die Mechanismen der Anfallserzeugung, Epileptogenese und Behandlungsresistenz zu verstehen. Sie untersuchen die Funktion neuer krankheitsassoziierter Genvarianten, die an Epilepsien beteiligt sind. Ein Projekt im Labor von Dr. Esquerra ist die Identifizierung kleiner Moleküle, die die STXBP1-Funktion in einem Zebrafisch-Modell retten können.
Universität von Pennsylvania Perelman School of Medicine
Dr. Benjamin Prosser ist Elternteil eines Kindes mit einer STXBP1‐Störung. Er arbeitet mit Dr. Beverly Davidson und Dr. Helbig, beide CHOP, zusammen. Gemeinsam arbeiten sie an neuen genetischen Therapien für STXBP1-Erkrankungen. Sie verwenden Anti-Sense-Oligonukleotiden (ASOs) zur Korrektur von Defiziten in der STXBP1-Genexpression. Sie testen vielversprechende Substanzen in iPSCs, die von STXBP1 Patienten generiert wurden. Ben Prossers Forschung über die Verwendung von Anti-Sense-Oligonukleotiden (ASOs) und miRNA zur Korrektur von Defiziten in der Stxbp1-Genexpression ist vielversprechend.
Weill Cornell Medizin
Das Labor von Dr. Burré untersucht pathologische Ereignisse an der neuronalen Synapse. Das Burré‐-Labor hat In‐vitro‐Modelle einschließlich iPSC‐Zellmodelle entwickelt, um STXBP1 zu untersuchen. Ihr Labor hat potenzielle Therapiekandidaten identifiziert. "Molekulare Chaperone" stellen die Struktur des STXBP1‐Proteins wieder her. Dise Studie ist ein niedermolekularer Ansatz zur Rettung der Proteinfehlfaltung bei kindlichen epileptischen Enzephalopathien. Für eines dieser kleinen Moleküle, 4-Phenylbutyrat, gibt es eine klinische Pilotstudie. Siehe auch "Breaking News".
ENGIN-Klinik am Kinderkrankenhaus von Philadephia (CHoP)
ENGIN − Epilepsy Neurogenetics Initiative − am Children's Hospital of Philadelphia (CHoP) integriert genetische Tests von Kindern mit schweren oder ungeklärten Epilepsien in die Diagnose und Behandlung der Kinder. ENGIN ist eines der größten Zentren weltweit für die Versorgung von Kindern mit STXBP1 Gen Mutationen.
Weitere Information über Engine finden Sie hier (Englisch): Engine Klinik (CHop)
Studie für erwachsene Patienten am Kinderkrankenhaus von Philadelphia und an der Universität von Pennsylvania
Ärzte und Kollegen in Europa führen gemeinsam eine Beobachtungsstudie an Erwachsenen im Alter ab 18 Jahren mit STXBP1 Gen Mutationen durch. Das Ziel der Studie ist es, das gesamte Spektrum der Symptome mit denen Erwachsene mit STXBP1 konfrontiert sind, herauszufinden.
Epilepsie-Genetik Forschung an der Toronto Western Kilink
Dr. Danielle Andrade führt eine Studie über die Folgen entwicklungsbedingter und epileptischer Enzephalopathien bei Erwachsenen durch. Für die Studie werden Patienten ab 18 Jahren mit einer pathogenen Mutation in STXBP1 rekrutiert. Dr. Andrade führt ebenfalls eine Studie über den Übergang vom pädiatrischen zum erwachsenen Gesundheitssystem bei Patienten mit Epilepsie durch. Teilnehmen können Familien von Patienten ab 12 Jahren.
Fendeep Studie
In Spanien findet eine nicht kontrollierte klinische Pilotstudie mit dem Medikament Fenfluramin statt. Fenfluramin wird getestet an Patienten im Alter von 2 bis 35 Jahren mit entwicklungsbedingten und epileptischen Enzephalopathien (DEEs).
Weitere Information über diese Pilotstudie finden Sie hier (Englisch): Fenfluramin
Pazifik Studie
In USA wird ein Prüfpreparat namens LP352 von der Firma Longboard Pharmaceuticals zur Behandlung von Anfällen im Zusammenhang mit entwicklungsbedingten und epileptischen Enzephalopathien (DEEs) getestet. Es werden Patienten im Alter von 12 bis 65 Jahren aufgenommen.
Weitere Information über diese Studie finden Sie hier (Englisch): LP352 Studie
Natural History Studies:
1. STARR Studie ab Juli 2023
STARR ist eine neue mehrjährige Studie, die die Teilnehmer in den USA über einen längeren Zeitraum hinweg begleiten wird, um ein umfassendes Verständnis ihrer Entwicklung zu erhalten. Diese Daten können in klinischen Studien für STXBP1-bedingte Erkrankungen eingesetzt werden.
Die STARR-Studie wird zunächst an vier klinischen Zentren durchgeführt:
-Children's Hospital of Philadelphia,
-Children's Hospital Colorado,
-Baylor College of Medicine / Texas Children's Hospital
-Weill-Cornell in New York.
Die STARR-Studie ergänzt das Protokoll der STXBP1-Studie zur natürlichen Entwicklung, die vom Europäischen STXBP1-Konsortium (ESCO) in Europa durchgeführt wird.
Ingo Helbig, MD, ist der leitende Prüfarzt der Studie an mehreren Standorten. James Goss, PhD, wissenschaftlicher Direktor der STXBP1 Foundation, und Michael Boland, PhD von der University of Pennsylvania, sind die administrativen Leiter der Studie.
STARR Studie
2. Kooperation STXBP1 Foundation mit Ciitizen
Ciitizen ist eine neue STXBP1‐Natural‐History‐Studie. Diese Studie sammelt Krankenakten von mehreren Krankenhäusern und Ärzten, bei denen ein STXBP1 Patient gesehen wurde, um zu erfahren, wie sich eine STXBP1‐Gen Mutation auf das Leben einer Person auswirkt. Diese Informationen können die Forschung beschleunigen. Aber Arzneimittelforscher können nicht auf die Informationen, die sie benötigen, zugreifen. Aber Patienten können ihre Krankenakten entsperren und zur Forschung beitragen. In Amerika werden 150 Teilnehmer bis zum Herbst 2021 rekrutiert. Es handelt sich um eine globale Studie. Aber zurzeit können nur medizinische Aufzeichnungen in englischer Sprache angenommen werden. In der Zukunft werden weitere Sprachen unterstützt werden.
3. Simons Searchlight
Die STXBP1 Foundation hat eine gemeinsame Partnerschaft mit Simons Searchlight und Ciitizen. Simons Searchlight Studie wird von der Simons Foundation finanziert. Simons Searchlight sammelt Informationen über den Verlauf von Patienten mit spezifischen genetischen Veränderungen, die einen Zusammenhang mit neurologischen Entwicklungsstörungen haben, wie z.B. Epilepsie, Entwicklungsverzögerungen und Merkmale von Autismus einschließlich STXBP1.
4. Encoded Therapeutics
Encoded Therapeutics hat ein Forschungsprojekt namens STXBP1 ENGAGE gestartet, um mehr über Kindern mit STXBP1-RD zu erfahren. Mehrere deutsche Familien haben an der Umfrage teilgenommen.
5. Natural History Study in Deutschland
Die Nature History Study in Deutschland wurde beendet und die Ergebnisse werden demnächst veröffentlicht werden. Deutschland (Dr. Syrbe ‐ Heidelberg) war in einer Studie involviert und die deutschen STXBP1 Kinder wurden in dieser "Natural History Study" aufgenommen. Die Studie war eine Zusammenarbeit zwischen Philadelphia (US), Amsterdam, Dänemark und Deutschland. Eine "Natural History Study" untersucht Symptome und das Fortschreiten der Krankheit durch Beobachtung. Studien zum natürlichen Krankheitsverlauf und andere Projekte sind entscheidend für die Planung erfolgreicher klinischer Studien. Umfragen zum Verständnis der Auswirkungen und Prioritäten von Patienten und Familien sind wichtige Beiträge zur Vorbereitung klinischer Studien.
6. Patienteregistrierung in Kanada
An der University of British Columbia arbeitet Dr.Cyrus Boelman eng mit seinen Kollegen Dr. Jennifer Engle, Danielle Andrade und Cecil Hahn an einem kanadischen nationalen Register für STXBP1 zusammen. Sie befragen Menschen mit STXBP1-Erkrankungen in ganz Kanada.
Information zu dem Register finden Sie hier (Englisch): Patientenregister Kanada
7. COMBINEDBrain
Die Duke Universität und COMBINEDBrain haben einen FDA-Zuschuss bekommen, um eine Studie mit einem Instrument zur Messung der Kommunikationsfähigkeit, die sogenannte Observer‐Reported Communication Ability (ORCA)‐Skala, durchzuführen. Die ORCA‐Skala ermöglicht, die grundlegenden Kommunikationsfähigkeiten der SIXBP1 Kinder genauer zu beschreiben. Es können Veränderungen in der Kommunikation während einer klinischen Studie gemessen werden. Die ORCA‐Skala wird in der klinischen STXBP1 4‐Phenylbutyrat‐Studie eingesetzt.
Charlene Son Rigby, die Präsidentin und Mitbegründerin der STXBP1 Foundation, wird so bald wie möglich mit der Implementierung des ORCA in von der STXBP1 Foundation unterstützten klinischen Studien und Natural History Studies beginnen.
Information über ORCA finden Sie hier (Englisch): Observer-Reported Communication Ability (ORCA) Measure
CVI=Cortical Visual Impairment
Es gibt STXBP1 Kinder bei denen CVI diagnostiziert wird. CVI resultiert aus einer Schädigung der zerebralen visuellen Verarbeitungsstrukturen aufgrund einer Störung des visuellen Inputs im Gehirn. Derzeit gibt es nur begrenzte Kenntnisse über die zerebrale Wahrnehmungsstörung, um die Entwicklung einer pädiatrischen CVI vorherzusagen. Die genetischen Ursachen für CVI sind heute noch weitgehend unbekannt. CVI kann nicht mit einer Brille korrigiert werden. Denn die Augen können sehen, aber das Gehirn kann die visuelle Welt nicht interpretieren.
CVI erkennen
-Kinder mit CVI starren oft auf Licht. Man kann sie dabei beobachten, wie sie lange Zeit aus dem Fenster schauen oder auf eine Deckenleuchte starren. Es kann auch so aussehen, als ob sie auf Dinge schauen, die nicht da sind.
-Vertraute Objekte erkennen Kinder mit der Zeit, aber sobald diese aus dem Zusammenhang gerissen werden, ergeben keinen Sinn mehr.
-Die Kinder bevorzugen konsequent Spielzeug in einer Lieblingsfarbe, oft rot und gelb.
-Damit das Kind ein Spielzeug anschaut, muss es vor den Augen bewegt oder geschüttelt werden und es muss sich im Gesichtsfeld befinden.
-Das Kind braucht lange um etwas, was ihm gezeigt wird, zu finden (Visuelle Latenzzeit).
-Bei großen Menschenmengen, wie in einem Warenhaus, schaltet das Kind ab, weil die visuelle Komplexität Schwierigkeiten bereitet.
-Kinder schauen nicht auf Spielzeug in der Ferne, sondern nur auf Spielzeug, das sich in der Nähe befindet.
-Es zeigen sich beim Kind atypische Sehreflexe. Es blinzelt, wenn eine Hand in Richtung des Nasenrückens bewegt wird.
-Gleichzeitig Schauen und Greifen funktioniert nicht. Das Kind schaut ein Spielzeug an, wendet sich ab und greift dann erst nach dem Spielzeug.
Die deutsche Übersetzung (pdf) der Präsentation von Magan Abbott über CVI auf dem 1. Europäischen Gipfel in Milan am 17. Mai 2023 können
Sie hier downloaden: Deutsche Übersetzung des Videos CVI
(Credit Daten und Grafik: STXBP1disorders.org)
Englisches Video: EU Summit in Milan
16. Was ist ein EU Register/Natrural History Study? Was ist ESCO? |
EU Register/Natural History Study
Das europäische Register ist eine Datenbank mit anonymisierten elektroklinischen und paramedizinischen Daten von Patienten mit einer STXBP1-Variante (Mutation).
Das Ziel des Registers, das in Dänemark gehostet wird, ist das Verständnis der Krankheit zu verbessern, neue therapeutische Lösungen zu entwickeln und die Lebensqualität der Patienten zu verbessern.
Die Sicherheit der Daten wird ständig überprüft.

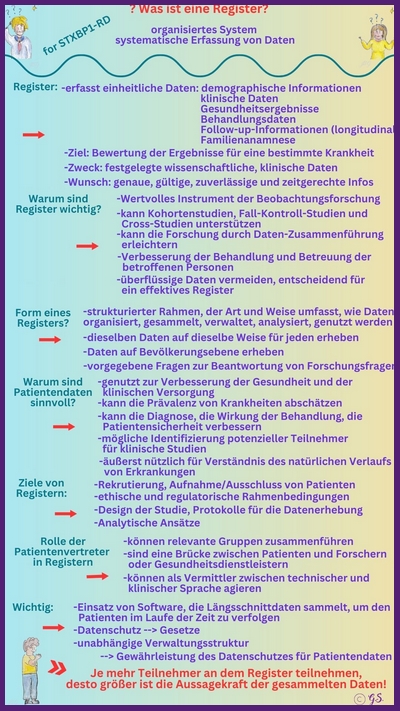
Copyright Gilberte Schnur
Zurzeit nehmen folgende Länder der EU am Register teil:
Dänemark, die Niederlande, Belgien, Deutschland, Frankreich, Italien, Spanien, Israel. Weitere Länder werden noch folgen.
Infos und Downloads auf folgender Webseite:
Webseite Register Heidelberg
Weitere Infos auf der ESCO Webseite:
ESCO Register
Natural History Study
ESCO wird eine groß angelegte, europaweite Studie zum natürlichen Krankheitsverlauf von STXBP1-Erkrankungen (STXBP1-RD) durchführen
In der Studie werden Teilnehmer unterschiedlichen Alters rekrutiert, um eine umfassende Repräsentation der STXBP1-Population in verschiedenen Lebensphasen zu gewährleisten. Die Teilnehmer werden an teilnehmenden Standorten aufgenommen und engmaschig überwacht. Diese Studie ist eine nicht-interventionelle Studie, als klinischer Versuch konzipiert.
Die Studie wird mit 50 Patienten aus den acht Mitgliedsländern beginnen: den Niederlanden, Spanien, Belgien, Frankreich, Deutschland, Dänemark, Israel und Italien.
Die Natural History Study wurde auf der Webseite NIH (Natural Library of Medicine) veröffentlicht:
Clinical Trials
Weiter Infos auf der ESCO Webseite:
ESCO Natural History Study


Copyright Gilberte Schnur
ESCO
Die Entwicklung eines neuen europäischen STXBP1-Konsortiums (ESCO) wurde schon von Dr. Ganna Balagura von der VU Amsterdam und der Universität Genua auf dem Summit 2022 erörtert.
Die große Neuigkeit der ersten europäischen Konferenz in Milan, im Mai 2023, war, dass das ESCO-Konsortium offiziell gegründet wurde. Das ist das erste europä ische STXBP1-Konsortium, das offiziell seine Arbeit aufnimmt und ein Patientenregister sowie eine Studie zum natürlichen Krankheitsverlauf einrichten wird. Die Studie über den natürlichen Verlauf wird zunächst neun Länder, wie oben schon erwähnt, umfassen, darunter die Niederlande, Dänemark, Belgien, Frankreich, Spanien, Deutschland, Italien und Israel, mit dem Ziel, zwei weitere europäische Länder einzubeziehen.
Ziel des ESCO ist es, die klinische und präklinische Forschung zu STXBP1 zu fördern und die Diagnose und klinische Bewertung in mehreren europäischen Ländern zu synchronisieren, damit STXBP1-Patienten unabhängig von ihrem Wohnort auf die gleiche Weise medizinisch betrachtet und bewertet werden.
Das ESCO-Konsortium, besteht aus Ärzt⋆innen und Forscher⋆innen und wurde nun durch Patientenvertreter⋆innen (1 Person pro EU Land) ergänzt.
Diese Person bespricht mit dem Konsortium, ob die Aktivitäten den Bedürfnissen der Familien entsprechen.
Die neue STARR-Studie in den USA ergänzt das Protokoll der STXBP1-Studie zur natürlichen Entwicklung, die vom Europäischen STXBP1-Konsortium (ESCO) in Europa durchgeführt wird.
Eine der wichtigsten Botschaften, die die Forscher während der Konferenz mehrfach hörten, war die Beschleunigung der Forschung durch offene Zusammenarbeit, offenen Austausch und Veröffentlichung von Daten. Dies alles wird den Forschungsfortschritt beschleunigen.
Im September 2025 kommen Familien und Organisationen aus ganz Europa und Israel im Rahmen des STXBP1-Awareness Month zu einer einzigartigen Spendenaktion zusammen. Jedes Land veranstaltet seine eigene Spendenaktion – die meisten organisieren Staffelläufe der Hoffnung, um das Bewusstsein zu schärfen und bahnbrechende Forschungsarbeiten zu unterstützen. Infos auch unter "Awareness Month".
Webseite ESCO:
Webseite ESCO
17. STXBP1 Kinder in Deutschland |
Lorena, geb. am 01.02.2003

Das ist Lorena! (Urheberrecht Bild: Snezana Adulovic)
Lorena ist aktuell 16 Jahre alt und mitten in der Pubertät. Sie ist ein geselliges und fröhliches Mädchen. Gerne würde sie sich in diesem Alter ein bisschen von den Eltern abnabeln, aber ihr seltener Gendefekt STXBP1 schränkt sie ein und sie ist über den kompletten Tag auf Hilfe angewiesen. Das macht sie manchmal wütend. Lorena geht in eine Körperbehindertenschule und sie freut sich immer sehr ihre Freunde dort zu treffen.
Lorena kam als gesundes Mädchen zur Welt, so dachten wir und die Ärzte. Die Schwangerschaft und Geburt verlief normal. Mit ca. 3 Monaten bemerkten wir bereits wie ihr Köpfchen wackelte, der Rumpf hatte wenig Stabilität. Bei den U Untersuchungen sprachen wir das an, aber es hieß, sie ist noch klein, das kommt noch. Allerdings mit 6 Monaten sahen auch die Ärzte, dass hier etwas nicht stimmte. Sie schaffte es lediglich das Köpfchen zu heben und sich seitlich zu rollen.
Es begannen etliche Untersuchgen wie MRT, Blutuntersuchen bei jeglichen Ärzten, Heilpraktikern, Osteopathen. Wir wussten uns nicht mehr zu helfen, keiner konnte helfen und wir waren heillos überfordert. Es begannen auch gleich etliche Förderungen 2 x wöchentlich Krankengymnastik (Vojta, Bobath), Frühförderung, schwimmen, später dann Ergotherapie und therapeutisches Reiten.
Lorena begann mit ca. 2,5 h Jahren zu Laufen. Sehr wackelig, durch die Ataxie, aber sie war stolz! Schon als Baby strampelte sie immer extrem viel mit den Beinchen. Zur selben Zeit etwa gingen wir auf eigene Kosten nach Bosten/USA an die Harvard University. Wir fühlten uns in Deutschland nicht gut betreut und keiner konnte uns sagen, was mit diesem immer lächelnden und fröhlichen Mädchen los ist. Alle Untersuchungen, die bereits in Friedrichshafen und Tübingen gemacht wurden, ließen wir übersetzen und schickten sie vorher an die Klinik in Boston; dort konzentrierten sich die Ärzte hauptsächlich auf genetische Untersuchungen. Und das erste Mal nahm sich dort ein Arzt endlich Zeit für mich, er erklärte mir eindringlich, dass unsere Tochter Lorena einen Gendefekt haben muss. Die globale Entwicklungsstörung belegt dies. Er erklärte mir, ich müsse mir das so vorstellen, wie wenn ich in eine Bibliothek komme und in irgendeinem Buch fehlt eine Seite. Diese Seite suchen wir jetzt bei ihrer Tochter. Ich glaube, dass ich mich dann das erste Mal damit abgefunden und es auch in irgendeine Weise angenommen habe.
Die Diagnose wurde aber trotzdem erst mit 13 Jahren gestellt. Lorenas Feinmotorik ist heute noch stark eingeschränkt, sie spricht wenige Wörter, ist zweisprachig aufgewachsen und versteht einfache Aufforderungen. Mit ihren Mitmenschen kann sie auf einfache Weise mit Hilfe von Gebärden (UK) kommunizieren.
Wir haben das große Glück, dass Lorena zu den wenigen gehört, die keine Epilepsie haben. Auch ansonsten ist sie organisch kerngesund und hat ein gutes Immunsystem. Sie entwickelt sich stets weiter, auf sehr langsamen Niveau
(Bild und Text veröffentlicht mit Genehmigung der Eltern.)
(Urheberrecht: Es ist strengstens untersagt, das Foto und/oder den Text zu downloaden, zu kopieren oder zu veröffentlichen!)
Lena-Marie, 27.04.2016 - 22.09.2024 |

Das ist Lena-Marie! (Urheberrecht Bild: Melanie Höfner)
Lena-Marie ist ein dreijähriges, sehr fröhliches und kontaktfreudiges Mädchen.
Tagtäglich zeigt sie uns, dass sie sich in ihrem Tempo weiterentwickelt. Sie kann alleine trinken und essen, auch wenn es unkonventionell aussieht.
Sie kann sich alleine hinsetzen und bewegt sich Popo rutschend, sehr zügig und mittlerweile auch zielgerichtet durch den Raum. Sie kommuniziert nonverbal, sowie mit wenigen doppelsilbigen Lauten. Lena-Marie mag Spielsachen mit Geräuschen und Lichteffekten.
Am 21.04.2017 erhielten wir die Diagnose Stxbp1.
Dem voran ging fast ein Jahr voller Vermutungen und Unsicherheit, warum Lena-Marie sich kaum motorisch, sowie kognitiv weiterentwickelte.
Im Alter von 2,5 Monaten bemerkten wir Zuckungen im Gesicht, die sich innerhalb von 3 Tagen zu ausgeprägten Krampfanfällen entwickelten. Nach einem einwöchigen Krankenhausaufenthalt bekamen wir die Diagnose Säuglingsepilepsie. Uns wurde gesagt, dass sich unser Kind wahrscheinlich normal weiterentwickelt, gegeben falls nur langsamer.
Jedoch wurde im Alter von 9 Monaten deutlich, dass sich Lena-Marie keinesfalls nur langsam weiterentwickelt. Wir suchten weitere Ärzte auf und bekamen kurz vor ihrem 1. Geburtstag die Diagnose "Stxbp1 mit epileptischer Enzephalopathie". Auf unsere Fragen hin "Was das bedeutet?", "Was das für Ausmaße haben wird?" usw., konnte man uns keine Auskünfte geben. Zu diesem Zeitpunkt hatte Lena-Marie dank täglicher Medikation keine epileptischen Anfälle mehr.
Aktuell besucht Lena-Marie einen integrativen Kindergarten. Sie ist ein voll akzeptiertes Mitglied in ihrer Gruppe, quietscht vor Freude und zeigt uns jeden Tag, wie schön das Leben sein kann.
(Bild und Text veröffentlicht mit Genehmigung der Eltern.)
(Urheberrecht: Es ist strengstens untersagt, das Foto und/oder den Text zu downloaden, zu kopieren oder zu veröffentlichen!)
Ema, geb. am 25.2.2015 |

Das ist Ema! (Urheberrecht Bild: Eva Auer)
Ema ist am 25.2.2015 in Salzburg geboren. Sie hat als gesundes Kind gegolten. Das aber etwas nicht stimmt, haben wir schon sehr früh erkannt. Sie hatte extremen Reflux, sie schlief bis zu 20 Stunden am Tag, sie konnte mit 4 Monaten immer noch nicht ihr Köpfchen halten. Die Entwicklung ist ins Stoppen geraten. Als sie genau 1 Jahr alt war, hat sie zum ersten Mal etwas in ihrer Hand halten können. Seitdem nimmt sie ständig, bis heute, alles in den Mund, muss ständig an etwas knabbern und beobachtet werden.
Mit 14 Lebensmonaten hatte sie für uns die ersten sichtbaren epileptischen Anfälle. Grand mall. Bis zu 15 Anfälle in 24 Stunden. Alle maximal bis zu 45 Sekunden lang. Meistens im Schlaf, oder in der Einschlafphase. Bei jedem Infekt, oder erhöhtem Fieber ab 38 Grad krampft sie. Auch zu wenig Schlaf oder zu viel Anstrengung führt zu Anfällen.
Mit 24 Monaten konnte sie sich selbst aufsetzen. Mit 36 Monaten konnte sie krabbeln. Mit 42 Monaten konnte sie laufen. Das alles nach Monaten langer Therapien und ständiger Übung verschiedener Methoden. Es fehlen ihr bis heute sämtliche Übergänge, das heißt, sie kann nicht frei aufstehen, sich nicht frei hinsetzen, etc. Das Laufen ist sehr hypoton.
Sie spricht nur sehr wenige Wörter, und mehrere Lauten. Wir wissen, dass sie uns versteht. Natürlich in ihrer Welt, und nur alltägliche Dinge. Wir fangen sehr bald mit Gebärdensprache an.
Sie ist ein glückliches, fröhliches Kind; in der Gesellschaft zurzeit gut integriert. Sie besuchte eine Krabbelgruppe und jetzt geht sie in den Kindergarten. Sie liebt Tiere, Kinder, Schwimmen und Musik. Sie kann mehrere Melodien einwandfrei summen.
Ihren geistigen Stand schätzen wir auf Grund Ihrer Handlung, Ihrer Spielzeug Wahl etc. auf maximal zwischen 12 bis 18 Monate je nach dem. Die Diagnose stxbp1 haben wir erst im Juni 2019 bekommen. Da war Ema schon 4,5 Jahre alt. Die Zusammenfassung ihrer Entwicklung: sie entwickelt sich ständig weiter, aber in extrem langsames Tempo. Ihre Epilepsie ist nach wie vor da.
(Bild und Text veröffentlicht mit Genehmigung der Eltern.)
(Urheberrecht: Es ist strengstens untersagt, das Foto und/oder den Text zu downloaden, zu kopieren oder zu veröffentlichen!)
Elena, 05.05.2004 - 15.05.2020 |
Die Erinnerung ist das einzige Paradies aus welchem wir nicht vertrieben werden können.
(Quelle: Jean Paul, Die unsichtbare Loge, 1793)
Zu Ehren unserer süßen Elena (Elli) Ramnialis (2004-2020), die plötzlich im Schlaf verstorben ist, bittet die STXBP1-Community Sie um eine Spende an "Elli's Smile Fund" zur Unterstützung der Forschung STXBP1 Disorders.
Hier können Sie spenden:
"Elli's Smile Fund"

Das ist Elena! (Urheberrecht Bild: Familie Ramnialis)
Sie ist heute 15 Jahre alt und mitten in der
Pubertät, wie jedes andere gesunde Mädchen in ihrem Alter. Aber Elli hat
STXBP1, einen sehr seltenen Gen-Defekt. Die Diagnose haben wir allerdings erst
bekommen, als Elena 12 Jahre alt war.
Die Geburt verlief normal, Elli kam laut den Ärzten als gesundes Mädchen zur Welt. So schien es und wir dachten es auch. Als jedoch die motorische Entwicklung lange auf sich warten ließ und sie sich einfach nicht zeitgemäß entwickelt hat, ahnten wir im Alter von 6-9 Monaten, daß irgendetwas nicht stimmte. Es folgten zahlreiche Arztbesuche, MRTs, Blutuntersuchungen ohne jegliche Diagnose.
Elena kam mit ca. 2,5 Jahren zum laufen, allerdings recht wacklig, da sie Ataxie hat. Das hat sich im Laufe der Jahre aber deutlich verbessert. Trotzdem ist die Feinmotorik sehr eingeschränkt. Auch heute noch. Da Elena wenig spricht, wenige zweisilbige Wörter; ist es schwierig einzuschätzen auf welchem geistigen Stand sie ist. Ich schätze zwischen 2-3 Jahren. Mit Hilfe von Gebärden (UK) kann sie mit ihren Mitmenschen auf einfache Art kommunizieren.
Wir haben das große Glück, dass Elli zu den 5 % gehört, die keine Epilepsie hat. Sie ist auch sonst ein organisch kerngesundes Mädchen mit einem richtig guten Immunsystem. Sie entwickelt sich stets weiter, aber auf sehr langsamen Niveau.
Elli liebt es Eis essen zu gehen, sie tanzt gerne und ist ein sehr fröhliches Kind. Sie besucht ein Körperbehindertenzentrum und geht sehr gerne in die Schule.
(Bild und Text veröffentlicht mit Genehmigung der Eltern.)
(Urheberrecht: Es ist strengstens untersagt, das Foto und/oder den Text zu downloaden, zu kopieren oder zu veröffentlichen!)
Eltern Blog |
Auf dem Blog der Vereinswebseite erfahren Sie, wie deutsche Familien von der STXBP1 Diagnose betroffen sind. Sie erfahren, wie die Kinder in ihrem Leben von der STXBP1 Diagnose eingeschränkt sind.
Für die Familien war es oft ein langer Prozess und viele Jahre der Ungewissheit, bis sie wussten, was ihrem Kind fehlt.
Eltern Blog

Geschrieben von Elenas Familie:
Unsere geliebte Tochter Elli ist am 15. Mai 2020 verstorben. Elli wurde am 5. Mai 2004 in Friedrichshafen geboren. Sie hatte eine seltene Mutation eines Gens namens STXBP1. Trotzdem war Elli immer glücklich, voller Freude und wusste, wie man das Leben genießt. Obwohl sie nur wenige Worte artikulieren konnte, war sie immer in der Lage, ihre Gefühle und auch ihre Wünsche nicht nur ihrer Familie, sondern der gesamten Umgebung, in der sie lebte, zu vermitteln.
Als die wenigen Wörter oder die Gebärdensprache, die sie benutzte, nicht ausreichten, um mit uns zu sprechen, drückte sie sich mit ihren schönen blauen Augen und ihrem Lächeln aus.
Elli liebte alle Arten von Musik, sie konnte den ganzen Tag tanzen. Wenn das Wetter es erlaubte, verbrachte sie Zeit entweder auf ihrer Schaukel im Garten oder in ihrem E-Bike und winkte und lächelte allen zu, die sie traf. Und ein guter Tag für Elli endete immer mit einer großen Portion Eis!
Elli hat uns in ihrem kurzen Leben so viele Dinge über Werte, Toleranz, Akzeptanz und Freude beigebracht. Da unser Leben von Ellis Bedürfnissen geprägt war, stehen wir jetzt vor einer großen Leere und einem großen Verlust und vermissen sie mehr, als Worte ausdrücken können.
Wir danken Ihnen allen für Ihre Spende an den "Elli's Smile Fund", der Sie bei der Suche nach einem Heilmittel für STXBP1-Erkrankungen unterstützt und anderen Kindern und Familien hilft.
Sonja & Christos (Urheberrecht Bild: Familie Ramnialis)
(Bild und Text veröffentlicht mit Genehmigung der Eltern.)
(Urheberrecht: Es ist strengstens untersagt, das Foto und/oder den Text zu downloaden, zu kopieren oder zu veröffentlichen!)
Die Eltern der STXBP1 Kinder in Deutschland haben eine WhatsApp Gruppe eröffnet und tauschen ihre Erfahrungen dort regelmäßig aus. In der Forschungsgruppe werden regelmäßig Updates zu aktuellen Forschungen veröffentlicht. |
Und plötzlich ist das Kind an einer seltenen, unheilbaren Krankheit erkrankt
Es kann nicht greifen, nicht krabbeln, nicht sitzen, nicht laufen, nicht sprechen wie andere Kinder; es entwickelt epileptische Anfälle. Über diesen Link kommen Sie auf die deutsche Vereinswebseite: |
Familientreffen im Rhön Park Aktiv Resort in Hausen-Rhön 2025
Vom 2. Oktober bis 5. Oktober 2025 treffen sich die deutschen STXBP1 Familien zum zweiten Mal im Rhön Park Aktiv Resort in Hausen-Rhön. Das Ziel des Familientreffens ist es neue Kontakte zwischen den betroffenen Familien aufzubauen.
Anmeldung bis zum 9. Februar 2025.

(Credit Grafik und Foto: STXBP1-ev.de)
Familientreffen im Rhön Park Aktiv Resort in Hausen-Rhön 2023
Vom 29. September bis 1. Oktober 2023 trafen sich die deutschen STXBP1 Familien zum ersten Mal. Das Ziel eines Familientreffens ist es Kontakte zwischen den betroffenen Familien aufzubauen. Der Informationsaustausch soll gefördert werden. Mehrere Familien reisten aus ganz Deutschland an.


(Credit Grafik und Foto: STXBP1-ev.de)
Erstes STXBP1 Familientreffen in Deutschland
Endlich war es am 29. September 2023 soweit, nach Monaten langen Vorbereitungen des STXBP1 e.V.
Am 29. September fuhren 21 STXBP1 Familien für einen Aufenthalt von 3 Tagen bis zu 8 Stunden mit dem Auto zum ersten deutschen STXBP1 Familientreffen in dem Rhön Park Hotel – Aktiv Resort in Hausen-Roth.



Was ein Wochenende.
Die meisten Familien kannten sich nur von der Whatsapp Gruppe und hatten sich noch nie live getroffen. Es war ein ganz besonderes Wochenende. Der persönliche Erfahrungsaustausch mit der STXBP1-RD-Krankheit, der auf diese Weise möglich wurde, war für alle ein ganz besonderes Erlebnis.
Das gemeinsame Frühstück, Mittag- und Abendessen ermöglichten viele intensive Gespräche.



Für Entspannung war für alle gesorgt: ein Bowlingabend für Eltern und Geschwisterkinder, gemeinsame Spaziergänge durch die schöne, erholsame Rhönlandschaft, fröhliche, lachende STXers mit Geschwistern in ihren Vereins-T-Shirts mit dem niedlichen Löwen im Rollstuhl und Hasen, spielerischer Spaß auf dem Trampolin oder der Rutsche im Indoor- oder Outdoor-Bereich des Hotels, nicht zu vergessen der Spaß im Schwimmbad und bei der Kinderdisco am Letzten Abend.



Bemerkenswert war die Beobachtung, wie die STXer miteinander kommunizierten. Ein 4-Jähriger stand vor einem 11-Jährigen im Rollstuhl und sie sahen sich lange und vertieft in die Augen. Ein anderer STXer streichelte zärtlich das Gesicht eines kleinen STXer-Mitkämpfers. Dann krabbelte der kleine Robber durch den Raum und fand es aufregend, im Kreis um einen kleinen Jungen zu krabbeln, der lachend auf dem Boden saß.
Fröhliche, glückliche, lachende, nonverbale STXBP1-Kinder, die keine Worte brauchen, um sich mitzuteilen, sondern sich mit Mimik, Körperberührung, Augenkontakt und Körpergesten unterhalten können. Eine wertvolle Erfahrung für die Eltern und ein Grund für den Verein, in Zukunft mehrere solche Familientreffen mit Spenden zu ermöglichen.
Dankbar für die Spenden und das gelungene Wochenende machten sich die 21 Familien am 1. Oktober 2024 wieder auf den langen Heimweg, mit viel Erfahrung im Gepäck, in der Hoffnung, sich bald wieder treffen zu können.
(Credit Fotos Eltern des STXBP1 e.V.)
English Version:
First STXBP1 Family Meeting in Germany
Finally, it was on 29 September 2024 so far, after months of long preparations of the STXBP1 e.V..
On September 29th, 21 STXBP1 families drove for a stay of 3 days up to 8 hours by car to the first German STXBP1 family meeting in the Rhön Park Hotel - Aktiv Resort in Hausen-Roth.
What a weekend.
Most of the families only knew each other from the WhatsApp group and had never met live. It was a very special weekend. The personal exchange of experiences with the STXBP1-RD disease, which became possible in this way, was a very special experience for everyone.
The common breakfast, lunch and dinner allowed many intensive conversations.
Relaxation was provided for everyone: a bowling evening for parents and siblings, walks together through the beautiful, relaxing Rhön landscape, happy, laughing STXers with siblings in their club T-shirts with the cute lion in the wheelchair and rabbit, playful fun on the trampoline or slide in the indoor or outdoor area of the hotel, not to forget the fun in the swimming pool and at the children's disco on the last evening.
It was remarkable to observe how the STXers communicated with each other. A 4-year-old stood in front of an 11-year-old in a wheelchair and they looked into each other's eyes for a long time, engrossed. Another STXer tenderly stroked the face of a little STXer fellow. Then the little robber, he crawled around the room, finding it exciting to crawl in circles around a little boy who was sitting on the floor, laughing.
Happy, joyful, laughing, non-verbal STXBP1 children who don't need words to communicate but can converse with facial expressions, body touch, eye contact and body gestures. A valuable experience for the parents and a reason for the association to facilitate several such family meetings in the future with donations.
Thankful for the donations and the successful weekend, the 21 families set off again on the long journey home on October 1, 2024, with a lot of experience in their luggage, hoping to meet again soon.
Weltweit haben etwa 1227 Kinder im Gen STXBP1 eine Mutation und sind krank, leiden u.a. an Epilepsie.
Bis jetzt gibt es keine Heilung für dieses STXBP1 Syndrom und die Medikamenten gegen Epilepsie helfen oft nicht.
In verschieden Ländern wird zurzeit geforscht, um eine Heilung für diese Kinder zu finden.
Helfen Sie, die STXBP1 Forschung voran zu treiben, denn diese Kinder brauchen eine Therapie!
Helfen Sie mit Ihrer Spende, die STXBP1-Krankheit besser zu verstehen und besser behandeln zu können.
Denn wissenschaftliche Forschung ist sehr teuer.
Sie können helfen:
Jede einzelne Spende ist wichtig, um STXBP1 Forschung zu unterstützen.
Und wie können Sie die STXBP1 Forschung fördern?
Klicken Sie auf den untenstehenden Link und spenden Sie.
Hier können Sie für z. B. die Forschung in Deutscland an den STXBP1 e.V. spenden:
STXBP1 e.V. Deutschland"
Sie können auch an die STXBP1 Stiftung in Amerika spenden.
Zweck der STXBP1 Foundation ist die Förderung der wissenschaftlichen Forschung auf dem Gebiet der STXBP1-Krankheit.
Hier können Sie an die STXBP1 Foundation spenden:
STXBP1 Foundation - eine "NON-Profit-Organisation"
Jede Spende ist willkommen. Jede Spende zählt! Jeder kleine Betrag ist eine große Hilfe.
DANKE!
The Million Dollar Bike Ride - Lulu's Crew am 12. Juni 2021
Deutsche Eltern liefen für die STXBP1 Forschung
Lulu's Crew - Spendenseite deutscher Eltern von Kindern mit STXBP1-Mutation
Lulu's Crew ist Teil des "Million Dollar Bike Ride", und verfolgt den Zweck für die STXBP1 Forschung zu sammeln.
STXBP1 Enzephalopathie
Die STXBP1-Mutation verursacht epileptische Enzephalopathien und damit verbundene neurodevelopmentale Störungen. Es handelt sich um eine sehr seltene schwerwiegende Erkrankung, die die wichtigsten Gehirnfunktionen betrifft. In Deutschland wurden bislang 84 Kinder mit einer STXBP1-Genmutation diagnostiziert. Sie sind zwischen 11 Monaten und 30 Jahre alt. Ihre Erkrankungen sind durch schwere geistige Behinderung, Epilepsie, Autismus, motorische und verhaltensbezogene Beeinträchtigungen gekennzeichnet. Viele von ihnen können nicht laufen und sind nonverbal. Sieben STX-Kinder sind in den letzten sechs Monaten in Amerika an einem SUDEP (plötzlich auftretender, ungeklärter Tod bei Epilepsie) gestorben, ein 16-jähriges Mädchen und ein fast 3-jä:hrigen Jungen starben in Deutschland.
Bisher gibt es keine Heilung, so dass alle Hoffnungen in der Forschung liegen. Diese bietet vielversprechende Ansätze, ist jedoch zur Durchführung auf Spenden angewiesen.
Bitte unterstützen Sie die STXBP1-Forschung, damit den Erkrankten in Zukunft besser geholfen werden kann.
Für die betroffenen Kinder und Familien ist die Diagnose ein lebensveränderndes Ereignis, wie nachfolgenden Eltern Statements zeigen:
"Unser Kind leidet an Epilepsie. Er kann nicht sitzen, nicht laufen und nicht sprechen. Er wird über eine Magensonde ernährt."
"Wir kämpfen täglich mit epileptischen Anfällen trotz Vagus-Nerv-Stimulation und Medikamenten."
"Unser Kind hatte einen Anfall. Er hat am ganzen Körper gezittert. Er hat schnell geatmet. Sein Herz hat sehr schnell geschlagen. Sein Körper wurde total steif. Nach ein paar Sekunden war dann der Spuk vorbei."
"Als wir die Diagnose bekamen, brach die Welt für uns zusammen. Mittlerweile bewundere ich mein Kind jeden Tag aufs Neue. Sie bringt uns unheimlich viel bei und wir können so viel von ihr lernen."
"Man fängt an über Vieles nachzudenken, weil vielleicht andere Dinge wichtig werden. Man lebt viel bewusster."
"Unsere Kinder sind besonders. Sie verändern alles in unserem Leben und zeigen uns, was wichtig ist."
Radfahren in Deutschland zur Unterstützung der STXBP1-Forschung
Zur Unterstützung der STXBP1 Forschung haben Eltern der STXBP1-Kids daher am 12. Juni 2021 in ganz Deutschland mehr als 2074 km geradelt und / oder gewandert, und hofften, dass viele Menschen für die STXBP1-Forschung spendeten. Jede Spende wurde der STXBP1-Stiftung zuteil, die hiermit Forschung in Amerika und Europa unterstützt, damit alle STXkids Hilfe und Heilung erfahren.
Das haben wir gesammelt: Lulu's Crew - Spenden für die STXBP1 Forschung
DANKE!
Vereine aus DE, USA, NL, DNK, ES, IT, CA, ISR, FR, PL, CHN, SK, BR, AU,TR
1. STXBP1 Verein Deutschland
Neue Erkenntnisse zu angeborenen Epilepsien
Eine großzügige Förderung in Höhe von 310.000 Euro der Dietmar Hopp Stiftung ermöglichte es, Dr. Syrbe und sein Team ein Patientenregister für die genetischen Ursachen angeborener Epilepsien zu etablieren – ein Projekt, das für die medizinische Forschung und Behandlung dieser komplexen Erkrankungen von unschätzbarem Wert ist. Das grundlegende Verständnis der genetischen Faktoren, die zu angeborenen Epilepsien führen, ist essentiell, um effektive Therapiestudien zu entwickeln und durchzuführen. Mit der Förderung konnte eine solide Datenbasis erstellt werden, die nicht nur die Diagnose und Prognose verbessern wird, sondern auch maßgeschneiderte Behandlungsansätze ermöglicht. Die Schaffung dieses Registers ist ein entscheidender Schritt hin zu einer personalisierten Medizin, bei der Patienten mit seltenen genetischen Formen von Epilepsie Hoffnung auf spezifischere und wirksamere Therapien haben können.
Link:
Neue Erkenntnisse zu angeborenen Epilepsien bei Kindern
In Deutschland fand eine Natural History Study unter der Leitung von PD Dr. Med. Steffen Syrbe, Neuropädiatrie und prächirurgische Epilepsiediagnostik, Universität Heidelberg, und in Zusammenarbeit mit Kim Thalwitzer, statt. In dieser Studie wurden in Deutschland lebenden STXBP1 Kinder aufgenommen.
Das Ergebnis der Studie finden Sie hier:
Natural History and Developmental Trajectories of Individuals With Disease-Causing Variants in STXBP1
Das vor der Mitgliederversammlung aufgenommene Video können Sie sich anschauen:
Video hier publiziert mit Genehmigung von Dr. Syrbe und Kim Thalwitzer.
Was ist eine Natural History Study?
Eine naturhistorische Studie sammelt über eine Gruppe von Menschen, die an einer bestimmten Krankheit leiden − hier STXBP1 Disorders − Informationen, speziell über die Naturgeschichte einer Krankheit ohne Intervention, vom Ausbruch der Krankheit an.
Eine solche Studie sammelt Gesundheitsinformationen, um zu verstehen, wie sich der medizinische Zustand oder die Krankheit entwickelt und wie sie zu behandeln ist.
Kenntnis der Naturgeschichte einer Krankheit kann bei vielen Erkrankungen der Arzneimittelentwicklung zugutekommen. Das Ziel ist also ein geeignetes Medikament oder Therapie zu finden.
Die Studie verfolgt nicht nur den Verlauf von Krankheiten im Laufe der Zeit, sondern identifiziert demografische, genetische, umweltbedingte und andere Variablen, die den Arzneimittelentwicklungsprozess beeinflussen können.
Naturgeschichtliche Studien sind oft der erste Schritt bei der Entwicklung eines Forschungsprogramms für eine Krankheit.
Siehe auch:
Matthijs Verhage: Designing an STXBP1 Natural History Study. Professor Matthijs Verhage, PhD, ist Leiter der Abteilung für Funktionelle Genomik am VU University Medical Center in Amsterdam; und er arbeitet mit der STXBP1 Foundation zusammen.
Das Video des Webinars vom 11. September 2020 um 18 Uhr finden Sie hier:
Webinar Professor Verhage
2. STXBP1 Stiftung (USA)
 Quelle: Mit Genehmigung von "stxbp1disorders.org"
Quelle: Mit Genehmigung von "stxbp1disorders.org"
Die erste wissenschafliche STXBP1 Konferenz fand am 27. und 28. Juni 2019 in Philadelphia statt. Etwa 50 Familien mit STX-Kindern und 40 Forschern aus der ganzen Welt trafen sich um über die aktuellen Forschungen über die STXBP1 Krankheit zu imformieren. Es was ein interessaner Austausch zwischen Familien und Forschern.
Sie können sich alle damals "live" gesendete Videos hier noch "offline" anschauen:
SIFM 2019
Alle Sitzungs- und Umfrageergebnisse finden Sie hier (auf Englisch):
Umfrageergebnisse
Die STXBP1 Foundation arbeitet mit mehreren Forschern zusammen, um die Therapien für STXBP1 Kinder zu beschleunigen. Aktuelle STXBP1 Forschungsprojekte
Forscher aus Amsterdam zu Gast in Philadelphia (USA)
Dr. Mala Misra-Isrie und die Doktoranden Annemiek van Berkel und Hanna Lammertse aus Amsterdam (VU-Universität - STXBP1 Forschung Amsterdam) hatten die Gelegenheit, ihre Arbeit bei der ersten STXBP1 Konferenz in Philadelphia zu präsentieren.
Information ihrer Präsentation finden Sie hier (auf Englisch): Präsentation der Forscher aus Amsterdam
3. STXBP1 in den Niederlanden: VU-Universität Amsterdam
Die Webseite der VU-Universität Amsterdam wurde erstellt, um Hintergrundinformationen zur STXBP1-Enzephalopathie bereitzustellen.
Hier finden Sie (auf Englisch) weitere Informationen zu Symptomen, Ursachen und Forschungsarbeiten der VU-Universität Amsterdam:
STXBP1 Amsterdam
An der VU-Universität Amsterdam fanden 2 Forschungstage statt:
1. Forschungstag:
Am Samstag, den 13. Oktober 2018, kam die erste Gruppe von 8 STXBP1-E-Familien an die VU-Universität Amsterdam, um an der neuen Studie zur STXBP1-Enzephalopathie teilzunehmen.
Mehr Information über diesen ersten STXBP1 Forschungtag finden Sie hier (auf Englisch): 1. Forschungstag am 13.Oktober 2018
2. Forschungstag:
Sieben neue STXBP1-Patienten nahmen an dem 2. STXBP1-Forschungstag teil.
Mehr Information über diesen zweiten STXBP1 Forschungtag finden Sie hier (auf Englisch): 2. Forschungstag am 13. April 2019
4. STXBP1 in Dänemark
Am 2. September 2019 trafen sich Familien mit STX-Kindern und Forschern, die über die aktuellen Forschungen imformierten, in Dianalund, Dänemark.
Mehr Information über dieses Treffen finden Sie hier (auf Englisch): STXBP1 Meeting in Dänemark
5. STXBP1 Verein Spanien
Mehr Information über den spanischen Verein finden Sie hier (auf Spanisch): Spanische STXBP1 Webseite / Asociación Síndrome STXBP1
6. STXBP1 Verein Italien
Mehr Information über den italienischen Verein finden Sie hier (auf Italienisch): Italienische STXBP1 Webseite
7. STXBP1 Verein Kanada
Kanada führt ein nationales Patientenregister, um Menschen mit einer STXBP1 Gen Mutation zu befragen.
Mehr Information über diese Webseite finden Sie hier (auf Englisch/Deutsch): Kanadische STXBP1 Webseite
8. STXBP1 Vereine Israel
Rafa's Moonshot - Israel
Rafa's Moonshot ist ein israelischer, gemeinnütziger Forschungsfonds, der sich für das Verständnis und die Behandlung von STXBP1-bezogenen Erkrankungen einsetzt. Der Fonds wurde von den Eltern Sagi Gidali, einem Unternehmer im Bereich Cybersicherheit, und Ella Gordon, einer Neurologin, gegründet, nachdem bei ihrem Sohn Raphael STXBP1 diagnostiziert worden war.
Sie setzen ihre einzigartige Mischung aus medizinischem und unternehmerischem Fachwissen ein, um innovative Forschungsprojekte voranzutreiben, die weltweite STXBP1-Gemeinschaft zu unterstützen und die Zusammenarbeit mit führenden Wissenschaftlern und Institutionen zu fördern. Ihr Ziel ist es nicht nur, Raphael zu helfen, sondern auch allen, die von dieser seltenen genetischen Erkrankung betroffen sind, Hoffnung und Lösungen zu bieten.
Mehr Information über diese Webseite finden Sie hier: Rafa's Moonshot - Israel
STXBP1 Webseite für Israel - Rare Smile
Eine Brücke zwischen westlicher und östlicher Hemisphäre: die Webseite für Israel.
Mehr Information über diese Webseite finden Sie hier: STXBP1 Webseite Israel
9. STXBP1 Verein Frankreich
Mehr Information über den französischen Verein finden Sie hier (auf Französisch): Französische STXBP1 Webseite
10. STXBP1 Verein Polen
Mehr Information über den polnischen Verein finden Sie hier (auf Polnisch): Polnische STXBP1 Webseite
11. STXBP1 Verein China
Mehr Information über diesen Verein finden Sie hier (auf Chinesisch): Chinesische STXBP1 Webseite
Basen-editierte Cynomolgus-Affen imitieren Kernsymptome der STXBP1-Enzephalopathie
Generation of base-edited monkeys
12. STXBP1 Verein Slowakei
Mehr Information über diesen Verein finden Sie hier (auf Slowakisch): Slowakische STXBP1 Webseite
13. STXBP1 Verein Brasilien
Mehr Information über diesen Verein finden Sie hier (auf Portugiesisch): Brasilianische STXBP1 Webseite
14. STXBP1 Verein Australien
Mehr Information über diesen Verein finden Sie hier (auf Englisch): Australische STXBP1 Webseite
15. STXBP1 Türkei
Türkische Eltern von STXBP1-Kindern haben eine Seite eingerichtet, um ihre Sichtbarkeit zu erhöhen, vor allem, weil es im Internet fast keine türkischen Inhalte über STXBP1 gibt. Sie hoffen, dass neu diagnostizierte Familien sie auf diese Weise leichter finden können.
Sie ermutigen türkische Patienten, sich bei der STXBP1-FOUNDATION zu registrieren und sich in die STXBP1-Weltkarte einzutragen. Sie freuen sich neue Familien zu unterstützen, die sich an sie wenden.
Mehr Information finden Sie hier (auf Türkisch):
Türkische STXBP1 Gruppe
E-Mail Adresse: E-Mail senden (stxbp1.turkiye@gmail.com)
Internationale Zusammenarbeit
STXBP1 Foundation hat Partnerschaften mit mehreren Organisationen geschlossen, um die STXBP1 Forschung voranzutreiben:
1. CIITIZEN
Die STXBP1 Foundation ist eine Partnerschaft mit CIITIZEN eingegangen, um eine neue naturgeschichtliche Studie für STXBP1 zu entwickeln.
Erfahren Sie mehr: Citizen Natural History Study
2. ORCA
Die Duke University und COMBINEDBrain haben eine FDA-Förderung für ein Instrument zur Messung der Kommunikationsfähigkeit erhalten, das sogenannte Observer-Reported Communication Ability (ORCA) Measure.
STXBP1 ist eines der ersten Mitglieder von COMBINEDBrain, das die ORCA-Skala verwendet.
Erfahren Sie mehr: ORCA
3. EURORDIS
Die STXBP1 Foundation wurde als assoziiertes Mitglied von EURORDIS aufgenommen. Durch die Aufnahme bei EURORDIS wird die Präsenz von STXBP1-Enzephalopathie in Europa erheblich erweitert. Der internationale Rare Disease Day, der am 28. Februar gefeiert wird, wurde in 2008 von EURORDIS ins Leben gerufen.
Erfahren Sie mehr: Eurordis
4. RARE-X
Der gemeinsame Austausch von Patienteninformationen bei der Suche nach einer Heilung für STXBP1-Erkrankungen und andere seltene Krankheiten ist äußerst wichtig.
Die STXBP1 Foundation arbeitet mit RARE-X zusammen, um ein Datenerfassungsprogramm für die Familien aufzubauen.
Eine Teilnahme an diesem Datenerfassungsprogramm trägt dazu bei, die Forschung und Entwicklung neuer Medikamente, Geräte und anderer Therapien zu beschleunigen.
Erfahren Sie mehr;
RARE-X STXBP1 Disorders Data Collection Program
5. Global Genes
Global Genes und RARE - X haben fusioniert. Gemeinsam werden sie in der Lage sein, der nächsten Generation die nötigen Werkzeuge und Ressourcen zur Verfügung zu stellen, die benötigt werden, die Forschung und Therapieentwicklung voranzutreiben.
Erfahren Sie mehr:
Zusammenarbeit Global Genes un RARE-X>
6. RAREBASE
RAREBASE hat eine eine neurowissenschaftliche Wirkstoffforschungsplattform, die mit 15 Patientenorganisationen für seltene Krankheiten zusammenarbeitet, gestartet, um die Arzneimittelforschung bei mehreren Erkrankungen zu beschleunigen. Zu den Mitarbeitern dieser technologiegestützten Wirkstoffforschungsplattform gehört STXBP1 Research Foundation. Rarebase benutzt nicht länger die traditionelle Medikamentenentwicklungspipeline, sondern nutzt neue Technologien und wissenschaftliche Erkenntnisse.
Erfahren Sie mehr: RAREBASE - neurowissenschaftliche Wirkstoffforschungsplattform
7. Natural History Studies
Eine Natural History Study - eine naturkundliche Studie - ist eine Art Forschungsstudie, die untersucht, wie eine Krankheit im Laufe der Zeit auf natürliche Weise fortschreitet. Für die Forschung sind diese Studien äußerst wichtig. STXBP1 Patienten können an folgende Studien teilnehmen:
- SIMONS SEARCHLIGHT
- RARE-X
- CIITIZEN
- Capsida / Klinische Studie
Mehr Information: Natural History Studies
8. Erwachsenen Natural History STXBP1 Study
Die University of Pennsylvania und das Children's Hospital of Philadelphia (CHOP) führen zusammen mit Kollegen aus Europa eine Natural History Studie an STXBP1 Erwachsenen durch, um herauszufinden mit welchen Herausforderungen Erwachsene mit STXBP1 konfrontiert sind. Teilnehmen können STXBP1 Erwachsene im Alter von 18 Jahren oder älter.
Der Schwerpunkt folgender Studie liegt auf der klinischen Präsentation von STXBP1-DEE im Erwachsenenalter:
Erfahren Sie mehr:
Studie zum natürlichen Verlauf von STXBP1-Entwicklung im Erwachsenenalter
9. EpiCARE
Experten präsentieren eine Reihe von Webinaren über seltene und komplexe Epilepsien.
Die Teilnahme an diesen Webinaren ist kostenlos, eine Voranmeldung ist jedoch erforderlich. Die Themen beziehen sich auf die Diagnostik und Behandlung seltener und komplexer Epilepsien.
Erfahren Sie mehr:
EpiCARE Webinars
10. STXBP1 Global Connect
Gemeinsam sind wir stärker. Gemeinsam werden wir ein Heilmittel finden.
STXBP1 Global Connect ist ein Zusammenschluss von Gruppen aus der ganzen Welt, die sich alle zum Ziel gesetzt haben, das Leben von Patienten mit STXBP1-Erkrankungen zu verbessern.
STXBP1 Global Connect listet alle STXBP1-Organisationen auf, Websites, Gruppen in sozialen Medien oder E-Mail-Listen.
Erfahren Sie mehr: STXBP1 Global Connect
11. FDA - Food and Drug Administration
Am 12. April 2022 nahm die STXBP1-Stiftung zusammen mit Mitgliedern der STXBP1-Gemeinschaft an einer einstündigen Anhörung mit Vertretern der US-amerikanischen Food and Drug Administration (FDA) teil. Ziel war es, der FDA einen Überblick über das Leben von STXBP1-Betroffenen zu geben.
Erfahren Sie mehr:
Anhörung FDA
25. Elternperspektiven - "Du bist besonders und geliebt, weil du bist, wie du bist" |
Auf der Webseite des STXBP1 Vereins können Sie die bewegten Geschichten einiger Familien lesen:
Familiengeschichten aus Deutschland
Auch die STXBP1 Foundation Webseite hat einen Bereich für Familiengeschichten eröffnet. Dort gibt es eine wöchentliche Serie, die einen STX'er und seine Familie in den Mittelpunkt stellt, genannt STX Strong Sundays. Die bewegten Geschichten finden Sie auf "STX Strong Sundays (Englisch):
Familiengeschichten aus Amerika
In einem kurzen Dokumentarfilm werden einige amerikanische Familien vorgestellt. Sie sind stolz darauf ihre Erfahrungen mit der STXBP1 Gemeinschaft zu teilen. Weitere Filme können auf dem Disorder Channel angeschaut werden. Übersetzungsmöglichkeiten bietet die "English Version" der Webseite:
Dokumentarfilm
Von der Delphintherapie bis zur Flucht aus der Ukraine
Voller Hoffnung reist die Familie Zuzkovicova aus der Slowakei nach Odessa in der Ukraine.
Die bewegte Geschichte finden Sie als pdf-Datei auf Deutsch und Englisch:
Von der Delphintherapie zur Flucht aus Odessa
Die Geschichte der Emma Rose - USA, geb. am 26.8.2008
Emma Rose wurde am 26. August 2008 geboren. Erst am 18. Dezember 2013 erhielt die Familie die STXBP1 Diagnose, nach 5 ½ Jahren des Suchens nach der Ursache für Emmas Epilepsie und weitere Entwicklungsstörungen − Eine Diagnose, die das Leben einer Familie komplett verändert.
Emma Rose leidet an Epilepsie. Lesen Sie die bewegte Geschichte der Emma Rose, eine Geschichte (auf Englisch), die beispielhaft für fast alle etwa 1227 STX-kids auf der Welt steht − ein langer Weg des Kampfes in eine ungewissene Zufunft der Familien, die alle auf die Entdeckung der Forschung, einer Therapie für das STXBP1 Syndrom, hoffen.
Um die Privatsphäre der Familie zu schützen, müssen Sie sich zum Lesen dieser Geschichte mit Ihrer E−Mail Adresse anmelden:
(platzierter Link mit Genehmigung der Familie Clatterbuck − STXBP1 Foundation)
Emma Rose auf STXBP1 Foundation
SE-ATLAS: Versorgungsatlas für Menschen mit seltenen Erkrankungen
Der SE-ATLAS gibt einen Überblick über die Versorgungsmöglichkeiten für Menschen mit seltenen Erkrankungen in Deutschland.
Mehr Information über die STXBP1-Enzephalopathie und den Verein STXBP1 e.V. finden Sie hier: SE-ATLAS
Die Universität Leiden, Niederlanden, hat alle bekannten STXBP1 Mutationen in einer Datenbank zusammengefasst.
Mehr Information über diese Datenbank finden Sie hier (auf Englisch): LOVD STXBP1 Datenbank
Finden Sie Menschen mit STXBP1 mithilfe der Karte
Setzten Sie sich mit Menschen mit einer STXBP1 Mutation in Verbindung und teilen Sie Ihre Erfahrungen. Treten Sie der STXBP1 Community bei.
Mehr Information über diese STXBP1 Weltkarte finden Sie hier (auf Englisch, auch auf Deutsch): Disease Map STXBP1
Eltern mit einem STXBP1 Kind können sich auf der Weltkarte des STXBP1 Foundation auch eintragen.
Wenn Sie möchten, dass Ihr STXBP1-Patient auf der STXBP1-Weltkarte des STXBP1 Foundation vertreten ist, tragen Sie sich einfach in deren Kontaktliste auf der STXBP1 Foundation Webseite ein:
World Map STXBP1 Foundation
Liste deutscher Kliniken
Weitere Kliniken können hinzugefügt werden. Bitte unter folgender E-Mail-Adresse melden:
gilberte.schnur@web.de
|
Universitätsklinikum Heidelberg |
!!! Für unvollständige oder fehlerhafte Angaben wird keine Haftung übernommen !!!
Von Eltern empfohlenen Reha Kliniken für STXBP1 Behandlung
Weitere Kliniken können hinzugefügt weden. Bitte unter folgender E-Mail-Adresse melden:
gilberte.schnur@web.de
|
Schön Klinik Vogtareuth |
!!! Für unvollständige oder fehlerhafte Angaben wird keine Haftung übernommen !!!
STXBP1 - Genetische Beratungsinstitute
Weitere Institute können hinzugefügt weden. Bitte unter folgender E-Mail-Adresse melden:
gilberte.schnur@web.de
|
Klinikum Oldenburg Universitätsinstitut für Medizinische Genetik |
!!! Für unvollständige oder fehlerhafte Angaben wird keine Haftung übernommen !!!
Unter folgender E-Mail-Adresse bin ich erreichbar:
gilberte.schnur@web.deDatenschutzinformationen
Wenn Sie uns per E-Mail kontaktieren, verarbeiten wir Ihre personenbezogenen Daten nur, soweit an der Verarbeitung ein berechtigtes Interesse besteht (Art. 6 Abs. 1 lit. f DSGVO), Sie in die Datenverarbeitung eingewilligt haben (Art. 6 Abs. 1 lit. a DSGVO), die Verarbeitung für die Anbahnung, Begründung, inhaltliche Ausgestaltung oder Änderung eines Rechtsverhältnisses zwischen Ihnen und uns erforderlich sind (Art. 6 Abs. 1 lit. b DSGVO) oder eine sonstige Rechtsnorm die Verarbeitung gestattet.
Ihre personenbezogenen Daten verbleiben bei uns, bis Sie uns zur Löschung auffordern, Ihre Einwilligung zur Speicherung widerrufen oder der Zweck für die Datenspeicherung entfällt (z. B. nach abgeschlossener Bearbeitung Ihres Anliegens). Zwingende gesetzliche Bestimmungen – insbesondere steuer- und handelsrechtliche Aufbewahrungsfristen – bleiben unberührt.
Sie haben jederzeit das Recht, unentgeltlich Auskunft über Herkunft, Empfänger und Zweck Ihrer gespeicherten personenbezogenen Daten zu erhalten.
Ihnen steht außerdem ein Recht auf Widerspruch, auf Datenübertragbarkeit und ein Beschwerderecht bei der zuständigen Aufsichtsbehörde zu.
Ferner können Sie die Berichtigung, die Löschung und unter bestimmten Umständen die Einschränkung der Verarbeitung Ihrer personenbezogenen Daten verlangen.
Details entnehmen Sie unserer Datenschutzerklärung.
Tipp: Bitte lesen Sie den Disclaimer und die Datenschutzerklärung!
Haftung für Inhalte Als Diensteanbieter sind wir gemäß § 7 Abs.1 TMG für eigene Inhalte auf diesen Seiten nach den allgemeinen Gesetzen verantwortlich. Nach §§ 8 bis 10 TMG sind wir als Diensteanbieter jedoch nicht verpflichtet, übermittelte oder gespeicherte fremde Informationen zu überwachen oder nach Umständen zu forschen, die auf eine rechtswidrige Tätigkeit hinweisen. Verpflichtungen zur Entfernung oder Sperrung der Nutzung von Informationen nach den allgemeinen Gesetzen bleiben hiervon unberührt. Eine diesbezügliche Haftung ist jedoch erst ab dem Zeitpunkt der Kenntnis einer konkreten Rechtsverletzung möglich. Bei Bekanntwerden von entsprechenden Rechtsverletzungen werden wir diese Inhalte umgehend entfernen. Haftung für Links Unser Angebot enthält Links zu externen Websites Dritter, auf deren Inhalte wir keinen Einfluss haben. Deshalb können wir für diese fremden Inhalte auch keine Gewähr übernehmen. Für die Inhalte der verlinkten Seiten ist stets der jeweilige Anbieter oder Betreiber der Seiten verantwortlich. Die verlinkten Seiten wurden zum Zeitpunkt der Verlinkung auf mögliche Rechtsverstöße überprüft. Rechtswidrige Inhalte waren zum Zeitpunkt der Verlinkung nicht erkennbar. Eine permanente inhaltliche Kontrolle der verlinkten Seiten ist jedoch ohne konkrete Anhaltspunkte einer Rechtsverletzung nicht zumutbar. Bei Bekanntwerden von Rechtsverletzungen werden wir derartige Links umgehend entfernen. Urheberrecht Die durch die Seitenbetreiber erstellten Inhalte und Werke auf diesen Seiten unterliegen dem deutschen Urheberrecht. Die Vervielfältigung, Bearbeitung, Verbreitung und jede Art der Verwertung außerhalb der Grenzen des Urheberrechtes bedürfen der schriftlichen Zustimmung des jeweiligen Autors bzw. Erstellers. Downloads und Kopien dieser Seite sind nur für den privaten, nicht kommerziellen Gebrauch gestattet. Soweit die Inhalte auf dieser Seite nicht vom Betreiber erstellt wurden, werden die Urheberrechte Dritter beachtet. Insbesondere werden Inhalte Dritter als solche gekennzeichnet. Sollten Sie trotzdem auf eine Urheberrechtsverletzung aufmerksam werden, bitten wir um einen entsprechenden Hinweis. Bei Bekanntwerden von Rechtsverletzungen werden wir derartige Inhalte umgehend entfernen. Zusatzerklärung zu Urheberrrecht Die Inhalte dieser Seiten wurden mit größter Sorgfalt erstellt. Für die Richtigkeit und Aktualität können jedoch keine Gewähr übernommen werden. Werbung Die Kontaktdaten des Impressums zur gewerblichen Nutzung zu gebrauchen, ist ausdrücklich nicht erwünscht. |
Tipp: Bitte lesen Sie die Datenschutzerklärung!
|
1. Datenschutz auf einen Blick Allgemeine Hinweise Die folgenden Hinweise geben einen einfachen Überblick darüber, was mit Ihren personenbezogenen Daten passiert, wenn Sie diese Website besuchen. Personenbezogene Daten sind alle Daten, mit denen Sie persönlich identifiziert werden können. Ausführliche Informationen zum Thema Datenschutz entnehmen Sie unserer unter diesem Text aufgeführten Datenschutzerklärung. Datenerfassung auf dieser Website Wer ist verantwortlich für die Datenerfassung auf dieser Website? Die Datenverarbeitung auf dieser Website erfolgt durch den Websitebetreiber. Dessen Kontaktdaten können Sie dem Abschnitt "Hinweis zur verantwortlichen Stelle " in dieser Datenschutzerklärung entnehmen. Wie erfassen wir Ihre Daten? Ihre Daten werden zum einen dadurch erhoben, dass Sie uns diese mitteilen. Hierbei kann es sich z. B. um Daten handeln, die Sie in ein Kontaktformular eingeben. Andere Daten werden automatisch oder nach Ihrer Einwilligung beim Besuch der Website durch unsere IT-Systeme erfasst. Das sind vor allem technische Daten (z. B. Internetbrowser, Betriebssystem oder Uhrzeit des Seitenaufrufs). Die Erfassung dieser Daten erfolgt automatisch, sobald Sie diese Website betreten. Wofür nutzen wir Ihre Daten? Ein Teil der Daten wird erhoben, um eine fehlerfreie Bereitstellung der Website zu gewährleisten. Andere Daten können zur Analyse Ihres Nutzerverhaltens verwendet werden. Welche Rechte haben Sie bezüglich Ihrer Daten? Sie haben jederzeit das Recht unentgeltlich Auskunft über Herkunft, Empfänger und Zweck Ihrer gespeicherten personenbezogenen Daten zu erhalten. Sie haben außerdem ein Recht, die Berichtigung oder Löschung dieser Daten zu verlangen. Wenn Sie eine Einwilligung zur Datenverarbeitung erteilt haben, können Sie diese Einwilligung jederzeit für die Zukunft widerrufen. Außerdem haben Sie das Recht, unter bestimmten Umständen die Einschränkung der Verarbeitung Ihrer personenbezogenen Daten zu verlangen. Des Weiteren steht Ihnen ein Beschwerderecht bei der zuständigen Aufsichtsbehörde zu. Analyse-Tools und Tools von Drittanbietern Beim Besuch dieser Webseite wird Ihren Besuch gezählt. Das geschieht mit einem sogenannten Counter. Detaillierte Informationen zu diesem Counter finden Sie am Schluss dieser Datenschutzerklärung. 2. Hosting Wir hosten die Inhalte unserer Webseite bei folgenden Anbieter: Strato Anbieter ist die Strato AG, Otto-Ostrowski-Straße 7, 10249 Berlin (nachfolgend: „Strato“). Wenn Sie unsere Website besuchen, erfasst Strato verschiedene Logfiles inklusive Ihrer IP-Adressen. Auftragsverarbeitung Wir haben einen Vertrag über Auftragsverarbeitung (AVV) mit dem oben genannten Anbieter geschlossen. Hierbei handelt es sich um einen datenschutzrechtlich vorgeschriebenen Vertrag, der gewährleistet, dass dieser die personenbezogenen Daten unserer Websitebesucher nur nach unseren Weisungen und unter Einhaltung der DSGVO verarbeitet. 3. Allgemeine Hinweise und Pflichtinformationen Datenschutz Die Betreiber dieser Seiten nehmen den Schutz Ihrer persönlichen Daten sehr ernst. Wir behandeln Ihre personenbezogenen Daten vertraulich und entsprechend der gesetzlichen Datenschutzvorschriften sowie dieser Datenschutzerklärung. Hinweis zur verantwortlichen Stelle Die verantwortliche Stelle für die Datenverarbeitung auf dieser Website ist: Gilberte Schnur Telefon: +49 (0) 6854 1416 Verantwortliche Stelle ist die natürliche oder juristische Person, die allein oder gemeinsam mit anderen über die Zwecke und Mittel der Verarbeitung von personenbezogenen Daten (z. B. Namen, E-Mail-Adressen o. Ä.) entscheidet. Speicherdauer Soweit innerhalb dieser Datenschutzerklärung keine speziellere Speicherdauer genannt wurde, verbleiben Ihre personenbezogenen Daten bei uns, bis der Zweck für die Datenverarbeitung entfällt. Wenn Sie ein berechtigtes Löschersuchen geltend machen oder eine Einwilligung zur Datenverarbeitung widerrufen, werden Ihre Daten gelöscht, sofern wir keine anderen rechtlich zulässigen Gründe für die Speicherung Ihrer personenbezogenen Daten haben (z. B. steuer- oder handelsrechtliche Aufbewahrungsfristen); im letztgenannten Fall erfolgt die Löschung nach Fortfall dieser Gründe. Allgemeine Hinweise zu den Rechtsgrundlagen der Datenverarbeitung auf dieser Website Sofern Sie in die Datenverarbeitung eingewilligt haben, verarbeiten wir Ihre personenbezogenen Daten auf Grundlage von Art. 6 Abs. 1 lit. a DSGVO bzw. Art. 9 Abs. 2 lit. a DSGVO, sofern besondere Datenkategorien nach Art. 9 Abs. 1 DSGVO verarbeitet werden. Im Falle einer ausdrücklichen Einwilligung in die Übertragung personenbezogener Daten in Drittstaaten erfolgt die Datenverarbeitung außerdem auf Grundlage von Art. 49 Abs. 1 lit. a DSGVO. Sofern Sie in die Speicherung von Cookies oder in den Zugriff auf Informationen in Ihr Endgerät (z. B. via Device-Fingerprinting) eingewilligt haben, erfolgt die Datenverarbeitung zusätzlich auf Grundlage von §25 Abs. 1 TTDSG. Die Einwilligung ist jederzeit widerrufbar. Sind Ihre Daten zur Vertragserfüllung oder zur Durchführung vorvertraglicher Maßnahmen erforderlich, verarbeiten wir Ihre Daten auf Grundlage des Art. 6 Abs. 1 lit. b DSGVO. Des Weiteren verarbeiten wir Ihre Daten, sofern diese zur Erfüllung einer rechtlichen Verpflichtung erforderlich sind auf Grundlage von Art. 6 Abs. 1 lit. c DSGVO. Die Datenverarbeitung kann ferner auf Grundlage unseres berechtigten Interesses nach Art. 6 Abs. 1 lit. f DSGVO erfolgen. Über die jeweils im Einzelfall einschlägigen Rechtsgrundlagen wird in den folgenden Absätzen dieser Datenschutzerklärung informiert. Widerruf Ihrer Einwilligung zur Datenverarbeitung Viele Datenverarbeitungsvorgänge sind nur mit Ihrer ausdrücklichen Einwilligung möglich. Sie können eine bereits erteilte Einwilligung jederzeit widerrufen. Die Rechtmäßigkeit der bis zum Widerruf erfolgten Datenverarbeitung bleibt vom Widerruf unberührt. Widerspruchsrecht gegen die Datenerhebung in besonderen Fällen sowie gegen Direktwerbung (Art. 21 DSGVO) WENN DIE DATENVERARBEITUNG AUF GRUNDLAGE VON ART. 6 ABS. 1 LIT. E ODER F DSGVO ERFOLGT, HABEN SIE JEDERZEIT DAS RECHT, AUS GRÜNDEN, DIE SICH AUS IHRER BESONDEREN SITUATION ERGEBEN, GEGEN DIE VERARBEITUNG IHRER PERSONENBEZOGENEN DATEN WIDERSPRUCH EINZULEGEN; DIES GILT AUCH FÜR EIN AUF DIESE BESTIMMUNGEN GESTÜTZTES PROFILING. DIE JEWEILIGE RECHTSGRUNDLAGE, AUF DENEN EINE VERARBEITUNG BERUHT, ENTNEHMEN SIE DIESER DATENSCHUTZERKLÄRUNG. WENN SIE WIDERSPRUCH EINLEGEN, WERDEN WIR IHRE BETROFFENEN PERSONENBEZOGENEN DATEN NICHT MEHR VERARBEITEN, ES SEI DENN, WIR KÖNNEN ZWINGENDE SCHUTZWÜRDIGE GRÜNDE FÜR DIE VERARBEITUNG NACHWEISEN, DIE IHRE INTERESSEN, RECHTE UND FREIHEITEN ÜBERWIEGEN ODER DIE VERARBEITUNG DIENT DER GELTENDMACHUNG, AUSÜBUNG ODER VERTEIDIGUNG VON RECHTSANSPRÜCHEN (WIDERSPRUCH NACH ART. 21 ABS. 1 DSGVO). WERDEN IHRE PERSONENBEZOGENEN DATEN VERARBEITET, UM DIREKTWERBUNG ZU BETREIBEN, SO HABEN SIE DAS RECHT, JEDERZEIT WIDERSPRUCH GEGEN DIE VERARBEITUNG SIE BETREFFENDER PERSONENBEZOGENER DATEN ZUM ZWECKE DERARTIGER WERBUNG EINZULEGEN; DIES GILT AUCH FÜR DAS PROFILING, SOWEIT ES MIT SOLCHER DIREKTWERBUNG IN VERBINDUNG STEHT. WENN SIE WIDERSPRECHEN, WERDEN IHRE PERSONENBEZOGENEN DATEN ANSCHLIESSEND NICHT MEHR ZUM ZWECKE DER DIREKTWERBUNG VERWENDET (WIDERSPRUCH NACH ART. 21 ABS. 2 DSGVO). Beschwerderecht bei der zuständigen Aufsichtsbehörde Im Falle von Verstößen gegen die DSGVO steht den Betroffenen ein Beschwerderecht bei einer Aufsichtsbehörde, insbesondere in dem Mitgliedstaat ihres gewöhnlichen Aufenthalts, ihres Arbeitsplatzes oder des Orts des mutmaßlichen Verstoßes zu. Das Beschwerderecht besteht unbeschadet anderweitiger verwaltungsrechtlicher oder gerichtlicher Rechtsbehelfe. Recht auf Datenübertragbarkeit Sie haben das Recht, Daten, die wir auf Grundlage Ihrer Einwilligung oder in Erfüllung eines Vertrags automatisiert verarbeiten, an sich oder an einen Dritten in einem gängigen, maschinenlesbaren Format aushändigen zu lassen. Sofern Sie die direkte Übertragung der Daten an einen anderen Verantwortlichen verlangen, erfolgt dies nur, soweit es technisch machbar ist. Auskunft, Löschung und Berichtigung Sie haben im Rahmen der geltenden gesetzlichen Bestimmungen jederzeit das Recht auf unentgeltliche Auskunft über Ihre gespeicherten personenbezogenen Daten, deren Herkunft und Empfänger und den Zweck der Datenverarbeitung und ggf. ein Recht auf Berichtigung oder Löschung dieser Daten. Hierzu sowie zu weiteren Fragen zum Thema personenbezogene Daten können Sie sich jederzeit an uns wenden. Recht auf Einschränkung der Verarbeitung Sie haben das Recht, die Einschränkung der Verarbeitung Ihrer personenbezogenen Daten zu verlangen. Hierzu können Sie sich jederzeit an uns wenden. Das Recht auf Einschränkung der Verarbeitung besteht in folgenden Fällen:
Wenn Sie die Verarbeitung Ihrer personenbezogenen Daten eingeschränkt haben, dürfen diese Daten – von ihrer Speicherung abgesehen – nur mit Ihrer Einwilligung oder zur Geltendmachung, Ausübung oder Verteidigung von Rechtsansprüchen oder zum Schutz der Rechte einer anderen natürlichen oder juristischen Person oder aus Gründen eines wichtigen öffentlichen Interesses der Europäischen Union oder eines Mitgliedstaats verarbeitet werden. SSL- bzw. TLS-Verschlüsselung Diese Seite nutzt aus Sicherheitsgründen und zum Schutz der Übertragung vertraulicher Inhalte, wie zum Beispiel Bestellungen oder Anfragen, die Sie an uns als Seitenbetreiber senden, eine SSL- bzw. TLS-Verschlüsselung. Eine verschlüsselte Verbindung erkennen Sie daran, dass die Adresszeile des Browsers von „http://“ auf „https://“ wechselt und an dem Schloss-Symbol in Ihrer Browserzeile. Wenn die SSL- bzw. TLS-Verschlüsselung aktiviert ist, können die Daten, die Sie an uns übermitteln, nicht von Dritten mitgelesen werden. Widerspruch gegen Werbe-E-Mails Der Nutzung von im Rahmen der Impressumspflicht veröffentlichten Kontaktdaten zur Übersendung von nicht ausdrücklich angeforderter Werbung und Informationsmaterialien wird hiermit widersprochen. Die Betreiber der Seiten behalten sich ausdrücklich rechtliche Schritte im Falle der unverlangten Zusendung von Werbeinformationen, etwa durch Spam-E-Mails, vor. 4. Datenerfassung auf dieser Website Cookies Unsere Internetseiten verwenden so genannte „Cookies“. Cookies sind kleine Datenpakete und richten auf Ihrem Endgerät keinen Schaden an. Sie werden entweder vorübergehend für die Dauer einer Sitzung (Session-Cookies) oder dauerhaft (permanente Cookies) auf Ihrem Endgerät gespeichert. Session-Cookies werden nach Ende Ihres Besuchs automatisch gelöscht. Permanente Cookies bleiben auf Ihrem Endgerät gespeichert, bis Sie diese selbst löschen oder eine automatische Löschung durch Ihren Webbrowser erfolgt. Cookies können von uns (First-Party-Cookies) oder von Drittunternehmen stammen (sog. Third-Party-Cookies. Third-Party-Cookies ermöglichen die Einbindung bestimmter Dienstleistungen von Drittunternehmen innerhalb von Webseiten (z. B. Cookies zur Abwicklung von Zahlungsdienstleistungen). Cookies haben verschiedene Funktionen. Zahlreiche Cookies sind technisch notwendig, da bestimmte Websitefunktionen ohne diese nicht funktionieren würden (z. B. die Warenkorbfunktion oder die Anzeige von Videos). Andere Cookies können zur Auswertung des Nutzerverhaltens oder zu Werbezwecken verwendet werden. Cookies, die zur Durchführung des elektronischen Kommunikationsvorgangs, zur Bereitstellung bestimmter, von Ihnen erwünschter Funktionen (z. B. für die Warenkorbfunktion) oder zur Optimierung der Website (z. B. Cookies zur Messung des Webpublikums) erforderlich sind (notwendige Cookies), werden auf Grundlage von Art. 6 Abs. 1 lit. f DSGVO gespeichert, sofern keine andere Rechtsgrundlage angegeben wird. Der Websitebetreiber hat ein berechtigtes Interesse an der Speicherung von Cookies zur technisch fehlerfreien und optimierten Bereitstellung seiner Dienste. Sofern eine Einwilligung zur Speicherung von Cookies und vergleichbaren Wiedererkennungstechnologien abgefragt wurde, erfolgt die Verarbeitung ausschließlich auf Grundlage dieser Einwilligung (Art. 6 Abs. 1 lit. a DSGVO und §25 Abs. 1 TTDSG); die Einwilligung ist jederzeit widerrufbar. Sie können Ihren Browser so einstellen, dass Sie über das Setzen von Cookies informiert werden und Cookies nur im Einzelfall erlauben, die Annahme von Cookies für bestimmte Fälle oder generell ausschließen sowie das automatische Löschen der Cookies beim Schließen des Browsers aktivieren. Bei der Deaktivierung von Cookies kann die Funktionalität dieser Website eingeschränkt sein. Welche Cookies und Dienste auf dieser Website eingesetzt werden, können Sie dieser Datenschutzerklärung entnehmen. CCM19 Unsere Website nutzt CCM19, um Ihre Einwilligung zur Speicherung bestimmter Cookies auf Ihrem Endgerät oder zum Einsatz bestimmter Technologien einzuholen und diese datenschutzkonform zu dokumentieren. Anbieter dieser Technologie ist Papoo Software & Media GmbH, Auguststr. 4, 53229 Bonn (im Folgenden ”CCM19”). Wenn Sie unsere Website betreten, wird eine Verbindung zu den Servern von CCM19 hergestellt, um Ihre Einwilligungen und sonstigen Erklärungen zur Cookie-Nutzung einzuholen. Anschließend speichert CCM19 einen Cookie in Ihrem Browser, um Ihnen die erteilten Einwilligungen bzw. deren Widerruf zuordnen zu können. Die so erfassten Daten werden gespeichert, bis Sie uns zur Löschung auffordern, den CCM19-Cookie selbst löschen oder der Zweck für die Datenspeicherung entfällt. Zwingende gesetzliche Aufbewahrungspflichten bleiben unberührt. Der Einsatz von CCM19 erfolgt, um die gesetzlich vorgeschriebenen Einwilligungen für den Einsatz von Cookies einzuholen. Rechtsgrundlage hierfür ist Art. 6 Abs. 1 S. 1 lit. c DSGVO. Auftragsverarbeitung Wir haben einen Vertrag über Auftragsverarbeitung (AVV) mit dem oben genannten Anbieter geschlossen. Hierbei handelt es sich um einen datenschutzrechtlich vorgeschriebenen Vertrag, der gewährleistet, dass dieser die personenbezogenen Daten unserer Websitebesucher nur nach unseren Weisungen und unter Einhaltung der DSGVO verarbeitet. Anfrage per E-Mail, Telefon oder Telefax Wenn Sie uns per E-Mail, Telefon oder Telefax kontaktieren, wird Ihre Anfrage inklusive aller daraus hervorgehenden personenbezogenen Daten (Name, Anfrage) zum Zwecke der Bearbeitung Ihres Anliegens bei uns gespeichert und verarbeitet. Diese Daten geben wir nicht ohne Ihre Einwilligung weiter. Die Verarbeitung dieser Daten erfolgt auf Grundlage von Art. 6 Abs. 1 lit. b DSGVO, sofern Ihre Anfrage mit der Erfüllung eines Vertrags zusammenhängt oder zur Durchführung vorvertraglicher Maßnahmen erforderlich ist. In allen übrigen Fällen beruht die Verarbeitung auf unserem berechtigten Interesse an der effektiven Bearbeitung der an uns gerichteten Anfragen (Art. 6 Abs. 1 lit. f DSGVO) oder auf Ihrer Einwilligung (Art. 6 Abs. 1 lit. a DSGVO) sofern diese abgefragt wurde; die Einwilligung ist jederzeit widerrufbar. Die von Ihnen an uns per Kontaktanfragen übersandten Daten verbleiben bei uns, bis Sie uns zur Löschung auffordern, Ihre Einwilligung zur Speicherung widerrufen oder der Zweck für die Datenspeicherung entfällt (z. B. nach abgeschlossener Bearbeitung Ihres Anliegens). Zwingende gesetzliche Bestimmungen – insbesondere gesetzliche Aufbewahrungsfristen – bleiben unberührt. Quelle: https://www.e-recht24.de  |
Zusätzliche Datenschutzerklärung:
Besucherzähler von besucherzaehler-kostenlos.de
Diese Webseite verwendet einen externen Zähler, um die Anzahl der Webseitenaufrufe zu erfassen. Dafür wird ein Java-Script von einer externe Webseite geladen. Der Server von besucherzaehler-kostenlos.de speichert die IP-Adresse des Zugriffs anonymisiert und zeitlich begrenzt in einer LOG-Datei ab. Diese wird regelmä&sulig;ig unwiderruflich gelöscht.
Um die korrekte Funktionsweise des Zählers zu gewährleisten, speichert der Besucherzähler zudem einen sogenannten Session-Cookie auf dem Computer des Besuchers ab. Dieser wird in der Regel vom Browser gelöscht, sobald er geschlossen wird. In diesem Cookie werden keine persönlichen Informationen gespeichert. Er enthält lediglich die Information der aufgerufenen Domain, sowie einen boolschen Tag (true/false), um den Besucher als bereits gezählt zu markieren.
Es werden auch darüberhinaus keine persönlichen oder personenbezogenen Daten vom Besucherzähler erhoben. Eine Nachverfolgung oder Zuordnung der Zugriffe ist zu keiner Zeit möglich.
Ein besonderer Dank geht an www.urlaubsziel.info, durch dessen Unterstützung dieser kostenlose Service erst möglich gemacht wird.
Datenschutzerklärung für die Nutzung von Google Translate bzw. Google Übersetzer / Übersetzung
Die Besucher dieser Webseite haben die Möglichkeit diese Webseite in andere Sprachen durch Google Translator übersetzen zu lassen. Google Translator ist ein Drittanbieter und wird von Google Inc., 1600 Amphitheatre Parkway, Mountain View, CA 94043, USA betrieben. Wenn Sie bei Google Translator eine Sprache auswählen, wird Google Translator aktiviert und es werden Daten an Google übertragen. Auf die übertragenen Daten und Dauer der Speicherung (IP-Adresse und URL, Sprache und persönliches Surfverhalten) haben wir keinen Einfluss. Informationen zu der Datenerhebung von Google finden Sie auf Google Policies Privacy.
Wir übernehmen kein Gewähr für die falschen computergenerierten Übersetzungen durch Google Translator, die eventuell von den originalen Inhalten der Webseite abweichen und die manchmal nicht nur falsche, sondern auch gar beleidigende Ausdrücke enthalten können. Wir übernehmen keine Haftung für entstandene Schäden.
Tipp: Bitte lesen Sie den Disclaimer!
Webmaster :: Gilberte Schnur
Angaben gemäß § gemäß Paragraph 18 MSEV
Gilberte Schnur
Birkenweg 13
66649 Oberthal
Kontakt
Telefon: +49 (0) 6854 1416
Handy: +49 (0) 152 01998468
Telefax: +49 (0) 3212 2302146
E-Mail: gilberte.schnur@web.de
Redaktionell verantwortlich
Gilberte Schnur
Birkenweg 13
66649 Oberthal
Quelle: https://www.e-recht24.de

Tipp: Bitte lesen Sie den Disclaimer und die Datenschutzerklärung!
Bitte hier klicken:
Zur Laptop Version
© Gilberte Schnur 2019-2025





