Breaking News:
Gentherapie mit Capsid-002 / Pilot Project mit 4-Phenylbutyrat / iSNARE präklinische Therapiestudie
Breaking News: |
STXBP1 Foundation assoziiertes Mitglied bei Eurordis – Februar 2026
Die STXBP1-Stiftung wurde als assoziiertes Mitglied von EURORDIS im Februar 2026 aufgenommen.
EURORDIS gilt als die europäische Organisation, die sich auf die Bedürfnisse von Patienten mit seltenen Krankheiten spezialisiert hat, und findet in diesem Bereich große Beachtung. Diese Mitgliedschaft bei EURORDIS stärkt die wichtige Präsenz der STXBP1-Stiftung in Europa.
Kontakt:
STXBP1 Foundation neues Mitglied bei Eurordis
Mitgliedschaft Eurordis
Tevard Biosciences – ASGCT 2026 in Boston vom 11. bis 15. Mai 2026
Auf der ASGCT 2026 in Boston vom 11. bis 15. Mai 2026 stellte Tevard Biosciences seine firmeneigene "Suppressor-tRNA*"-Plattform vor.
Im Mittelpunkt ihrer Forschung steht die Entwicklung von Therapien zur Behandlung von Nonsense-Mutationen anstelle einzelner Gene. Bei ihrer Technik handelt es sich um eine "Read-Through"-Technik, was bedeutet, dass das Stoppcodon in einer Nonsense-Mutation außer Kraft gesetzt und erfolgreich umgangen werden kann, um die Produktion des Proteins in voller Länge wiederherzustellen.
Im Oktober 2023 erhielten sie von der STXBP1-Foundation eine Maus mit einer Nonsense-Mutation im STXBP1-Gen.
Eine Nonsense-Mutation verä,ndert ein einzelnes Nukleotid, also nur einen Buchstaben, in der DNA und es entsteht ein Stoppcodon in der mRNA-Sequenz, wodurch die Translation gestoppt wird und ein nicht-funktionsfähiges Protein entsteht.
Eine Nonsense-Mutation endet in einem genetischen Bericht immer mit einem X oder * oder TER. Dies symbolisiert ein vorzeitiges Stoppcodon. Beispiel im Bericht: p.Trp448&ast oder p.R235X oder GLn12 Ter
Tevard nutzt eine hochkompakte tRNA-Architektur, die in Adeno-assoziierte-Virus (AAV)-Vektoren verpackt ist, was eine präzise Dosierung und gewebespezifische Verabreichung ermöglicht. STXBP1 passt perfekt zur Plattform von Tevard, da es auf die Mutation abzielt und nicht nur auf das Gen.
Die Suppressions-tRNA-Programme von Tevard könnten das Potenzial haben, genetische Erkrankungen breit anzugehen. Das STXBP1-Nonsense-Mausmodell ist ein spezielles Werkzeug, das entwickelt wurde, um eine Nonsense-Mutation nachzubilden, die für etwa 23 % der menschlichen STXBP1-Erkrankungen verantwortlich ist. Die Nonsense-Mausmodelle ermöglichen es, therapeutische Plattformen wie die Suppressor-tRNAs von Tevard direkt in vivo (im Menschen) zu validieren.
Das STXBP1-Nonsense-Mausmodell besitzt genau diese durch ein Stoppcoden vorzeitig abgebrochene STXBP1-Gensequenz. Dieses STXBP1-Mausmodell wurde in Zusammenarbeit mit "The Jackson Laboratory" entwickelt und finanziell von "Clara Inspired" unterstützt, einer gemeinnützigen Patientenvertretungsorganisation, die die Entwicklung des Werkzeugs finanzierte, während die STXBP1-Foundation die Aufrechterhaltung des Ökosystems finanziert, damit das Werkzeug zur Suche nach einem Heilmittel genutzt werden kann.
*tRNA transportiert die passende Aminosäure zum Ribosom ("Proteinfabrik der Zelle"), damit ein Protein gebildet werden kann.
Kontakt:
Tevard tRNA-based therapies
Klinische Studie EMERALD
Relutrigin ist ein experimenteller Wirkstoff, ein erstklassisches kleines Molekül, das derzeit für die Behandlung bestimmter genetischen Epilepsieformen erforscht wird. Es wird auch eine mögliche Anwendung bei STXBP1-RD geprüft.
Relutriging wirkt als Präzisionsmodulator von Natriumkanälen. Natriumkanäle sind für die elektrische Aktivität von Nervenzellen wichtig. Relutrigin könnte dazu beitragen, die neuronale
Übereregbarkeit zu regulieren und somit Anfälle zu reduzieren. Die Anwendung von Relutrigin bei STXBP1-RD ist derzeit (Prax-562) noch ein Forschungsansatz und wird in klinischer Studie geprüft.
Relutrigin wurde in der EMBOLD-Studie zur Behandlung von Anfällen untersucht. Neben der Anfallskontrolle
wurde auch über Verbesserungen in anderen Bereichen wie Wachheit, Verhalten und Kommunikation berichtet. Die Embold-Studie der Praxis Precision Medicines Inc. ist eine multizentrische, doppelblinde,
placebokontrollierte, randomisierte Studie, die ein positives krankheitsmodifizierendes Potenzial in der Phase-2-Studie zeigte.
Die EMERALD-Studie ist eine Phase-3, randomisierte, placebokontrollierte Studie, die Wirksamkeit, Sicherheit, Verträglichkeit und Pharmakokinetik von Relutrigin, dasselbe Prüfpräparat wie in der EMBOLD-Studie, untersucht. Derzeit gibt es aktive Standorte in den Vereinigte Staaten, Brasilien und Australien, (keine Standorte in Europa verfügbar). Es kann zwischen einer Teilnahme zu Hause, in der Klinik oder einer Kombination aus beiden gewählt werden. Praxis Precision Medicines Inc. sponsert die EMERALD-Studie.
Kontakt:
Klinische Studie EMERALD
EMERALD-Studie Clinical Trial
Klinische Studie DEEpOCEAN in Deutschland – Rekrutierung beendet
Bexicaserin ist eine Substanz, ein Prüfpreparat, das als potenzieller Modulator im Nervensystem untersucht wird. Es wird vorrangig im Kontext von Neurotransmissionsregulation erforscht, doch der genaue zellulären Wirkmechanismus ist nicht eindeutig etabliert.
Bexicaserin könnte theoretisch Einfluss auf SNARE-Komplexe haben, wodurch das Gleichgewicht exzitatorische versus inhibitorische Signale verschoben werden könnte. Deshalb wird Bexicaserin auch im Zusammenhang mit STXBP1-RD (DEEs) untersucht, aber es ist noch kein zugelassenes Medikament. Es wurde von Longboard Pharmaceuticals entwickelt und von Lundbeck übernommen.
Bexicaserin ist Teil der DEEpOCEAN-Studie, einer Phase-3-Studie für DEEs zu denen auch STXBP1-RD gehört. In einer PACIFIC-Studie (Phase 1b/2a) wurde Bexicaserin (auch: LP352) bei DEE-Patient:innen untersucht, mit positiven Ergebnissen für motorische Anfälle.
Das Medikament zeigt ein günstiges Sicherheits- und Verträglichkeitsprofil. Häufige Nebenwirkungen waren Infekte der oberen Atemwege, verringerter Appetit und Müdigkeit.
Bexicaserin hat von der FDA eine Breakthrough Therapy Designation für DEEs erhalten. Es braucht weitere Studien (kontrollierte Phase-3- Studien), um Wirksamkeit, Sicherheit und Langzeitwirkung bei verschiedenen DEEs zu bestätigen.
Die DEEpOCEAN-Studie (Phase 3) schließt auch STXBP1-Patienten:innen ein und wird global vorangetrieben, und startete auch in Deutschland, mit Startdatum: 08.10. 2025.
Bei Interesse an einer Studienteilnahme kann man sich bei den deutschen Studienzentren (Bielefeld, Kiel, Frankfurt, Ravensburg, Radeberg oder Bonn) melden.
Kontakt:
Klinische Studie DEEpOCEAN in Deutschland
Clinical Trial DEEpOCEAN Info
iSNARE präklinische Therapiestudie
ERDERA hat 18 multinationalen Projekte bekannt gegeben.
Es gab eine Ausschreibung zum Thema "Präklinische Therapiestudien für seltene Krankheiten unter Verwendung kleiner Moleküle".
Diese 18 Projekte wurden mit einem Gesamtbudget von rund 29 Millionen Euro für eine Förderung ausgewählt. Transnationalen Konsortien werden von 29 nationalen und regionalen Förderorganisationen aus 23 Ländern unterstützt und von der Europäischen Kommission kofinanziert.
Jedes Projekt soll solide präklinische Erkenntnisse liefern, um künftige klinische Studien vorzubereiten.
iSNARE (Partner Verhage, Amsterdam UMC & VU Amsterdam et al. – ESCO Mitglieder) ist eins von den 18 Projekten.
iSNARE befasst sich mit SNAREopathien.
Das sind seltene neurologische Entwicklungsstörungen, die durch Mutationen in Genen, wie STXBP1, verursacht werden, die die synaptische Sekretion steuern.
Für die Studie werden von aus Patienten gewonnenen iPSC-Neuronen und SNAREopathie-Mausmodellen verwendet.
STXBP1-RD ist eine SNAREopathie. SNAREs (Soluble NSF Attachment Protein Receptors) sind für die Vesikelfusion in Neuronen und anderen Zellen unerlässlich. Wenn ihre Funktion beeinträchtigt ist, kann dies zu Störungen der neuronalen Kommunikation und des zellulären Transports führen, was eine Reihe klinischer Symptome zur Folge hat. Weitere Infos zu SNAREopathien auf dieser Webseite: Eltern, Infothek.
Weitere Infos zur Studie hier:
iSNARE präklinische Therapiestudie

Encoded Therapeutics treibt eine Gentherapie für STXBP1-RD voran
Encoded Therapeutics treibt eine Gentherapie für STXBP1-RD durch gezielte Vektorentwicklung voran. Es handelt sich um eine auf Neuronen ausgerichtete Gentherapie mit dem Ziel, mehrere Phänotypen von STXBP1-RD zu retten. Sie zeigt Fortschritte bei Mäusen und wird von nichtmenschlichen Primaten gut vertragen.
Es wird ein AAV9-Vektor verwendet, der auf das ZNS abzielen kann. Die Verabreichung erfolgt über eine intrazerebroventrikuläre (ICV) Route im Gehirn.
Es wurden neuartige Promotoren* entwickelt, die eine Verteilung und Expression des stxbp1-Gens in Gabaergischen und glutamatergen Neuronen ermöglichen. Diese Neuronen sind entscheidend für die Rettung des Phänotyps des STXBP1-Mangels. In anderen Neuronen sollte die Expression geringer sein. Die Expression in DRG-Zellen** wird ebenfalls detargettiert. Diese Zellen sind bekanntermaßen entscheidend für die Toxizität in Gentherapien.
Bei Mäusen zeigte die Gentherapie eine Rettung der Verhaltensphänotypen. Sie zeigten eine Verbesserung der motorischen und kognitiven Funktionen und eine Verringerung der Anfälle. Die Mausversuche wurden auch an nicht-menschlichen Primaten durchgeführt und zeigten Sicherheit und Wirksamkeit mit einer weit verbreiteten Vektorverteilung im ZNS, auch nach ICV-Verabreichung, sowie Sicherheit und Detargeting für DRG.
Es besteht die Hoffnung, dass diese Ergebnisse die weitere klinische Entwicklung dieser Vektoren zur Behandlung von STXBP1 unterstützen.
*In der Gentherapie sind Promotoren entscheidende DNA-Abschnitte, die wie Startknöpfe (der Promotor ist eine Sequenz stromaufwärts (vor des eigentlichen Gens) für ein therapeutisches Gen funktionieren, indem sie steuern, wann und wie stark das Gen abgelesen und ein Protein produziert wird.
**Nervenzellen in den Dorsal Root Ganglia (DRGs), den Spinalganglien – wichtigen Knotenpunkte für die Schmerzweiterleitung vom Körper zum Rückenmark.
Quelle: Neuron targeted disorders in mice and is well tolerated in nonhuman primates, R. Aeran et al., Molecular Therapy, 2025.
Weitere Infos hier:
Encoded Therapeutics
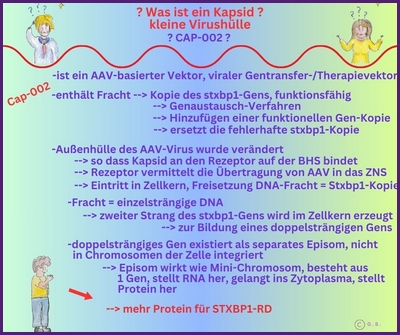
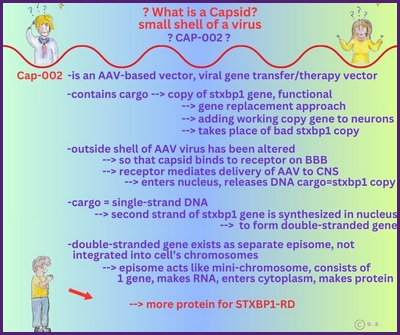
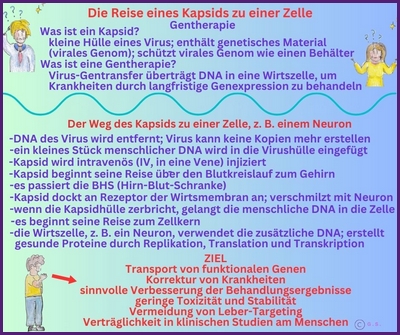
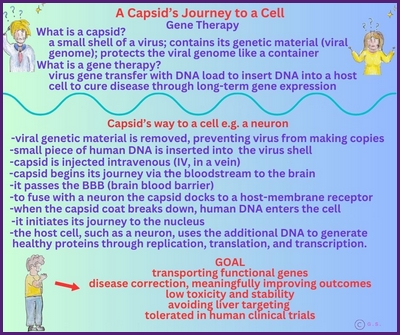
Gentherapie mit Capsid-002 von Capsida Biotherapeutics
Die Stxbp1 Community ist zutiefst erschüttert, mitteilen zu müssen, dass der erste Teilnehmer der klinischen Studie Capsida CAP-002 SYNRGY wenige Tage nach der Verabreichung verstorben ist. Unser tiefstes Mitgefühl und Beileid gilt der Familie, und wir bitten alle, in dieser äußerst schwierigen Zeit die Privatsphäre der Familie zu respektieren.
Weitere Infos hier:
Update zu Capsid-002 STXBP1 Foundation
Update zu Capsid-002 Capsida Biotherpeutics
Capsida
Biotherapeutics, ein Biotech-Unternehmen, hat bekannt gegeben, dass die US Food and Drug Administration
Arzneimittelbehörde (FDA) einen Orphan-Drug-Status für eine potenzielle Gentherapie für STXBP1-bezogene Erkrankungen erteilt hat.
Capsida führte diese Studien in Zusammenarbeit mit Mingshan Xue, Ph.D., außerordentlicher Professor für Neurowissenschaften und Molekular- und Humangenetik am Baylor College of Medicine, sowie Mitglied der Cain Foundation Laboratories und des Duncan Neurological Research Institute am Texas Children's Hospital durch. Eine klinische Studie mit Capsid-002 ist im dritten Quartal 2025 geplant. Die Rekrutierung hat in Philadelphia begonnen.
Alle Infos und Updates zu der klinischen Studie finden Sie hier:
Erste Gene Therapy für STXBP1-RD
Weitere Infos finden Sie auf folgenden Webseiten:
Orphan-Drug-Status Capsida
Pipeline for Rare and Common Diseases Capsida
Press Releases Capsida
Capsida Publications
Auch auf der STXBP1 Foundation Webseite gibt es ebenfalls die neuensten Informationen zu der klinischen Studie:
Oral presentation Capsida at ASGCT 2025
Copyright Gilberte Schnur
Preprint zu der Glycerinphenylbutyrat (Ravicti®) klinischen Studie wurde veröffentlicht.
Ravicti® hat weltweit den ersten Orphan-Status für ein Medikament gegen STXBP1 bekommen.
Weitere Infos auf:
Preprint klinische Studie für Ravicti®
Orphan Drug Status für Ravicti®
Immedica hat Glycerinphenylbutyrat (Ravicti®) bei STXBP1-bedingten Erkrankungen den Orphan-Status erteilt
Dies ist weltweit der erste Orphan-Status für ein Medikament gegen STXBP1!
Weitere Infos auf:
Orphan Drug Status für Ravicti®
Link zu Immedica (Vertrieb von Ravicti®):
Immedica gibt Orphan Drug Status für Ravicti® bekannt
Update Ergebnisse der 4-Phenylbutyrat Studie in New York
Forscher, u.a. Dr. Zachary Grinspan, vom Weill Cornell Medicine, zeigten auf dem 1. Europäischen Gipfel in Milam am 17. Mai 2023 weitere positive Ergebnisse der Ravicti Studie.
Sie können den Vortrag auf der Webseite der STXBP1 Foundation ansehen: EU Summit 2023 in Milan
Vidoes STXBP1 EU Summit 2023
Die deutsche Übersetzung (pdf) der Präsentation von Zach Grinspan über Ravicti auf der 1. Europäischen Gipfel in Milan am 17. Mai 2023 können
Sie hier downloaden: Deutsche Übersetzung des Videos Ravicti
(Credit Daten und Grafik: STXBP1disorders.org)
Erste Ergebnisse der 4-Phenylbutyrat Studie
Forscher, u.a. Dr. Zachary Grinspan, vom Weill Cornell Medicine, haben die ersten positiven Ergebnisse der Ravicti Studie gesammelt.
Die Ergebnisse wurden am 19. - 20. August 2022 auf der STXBP1 Summit+ in Philadelphia bekannt gegeben.
Sie können seinen Vortrag auf der Webseite der STXBP1 Foundation ansehen: Research Roundtable - Clinical Update
Vidoes STXBP1 Summit+ 2022
Hier mehr Info über die Studie (auf Englisch): Klinische Studie mit Glycerol Phenybutyrat
(Credit Daten und Grafik: STXBP1disorders.org)
Neue Therapie Studie für STXBP1 in US mit 4-phenylbutyrat
Forscher von Weill Cornell Medicine leiten die Entwicklung eines Pilot-Behandlungsprotokolls für STXBP1-Enzephalopathie mit Epilepsie mit einem finanziellen Zuschuss vom "The Orphan Disease Center" und "Clara Inspired".
In einer revolutionären Studie fanden Dr. Jacqueline Burré und ihre Kollegen heraus, dass ein Medikament namens Phenylbutyrat als chemisches Chaperon wirken kann, um das funktionelle STXBP1 zu stabilisieren. Diese Studie zielt darauf ab, Phenylbutyrat als Behandlungsoption in die klinische Praxis einzuführen.
Unter der Leitung von Dr. Zachary Grinspan, Direktor der Pädiatrischen Epilepsie, werden die Forscher von Weill Cornell Medicine zehn Kindern mit STXBP1-Enzephalopathie Phenylbutyrat verabreichen. Es werden auch Interviews mit Familienmitgliedern durchgeführt, um mehr über ihre Erfahrungen mit STXBP1 und mit dem Studienmedikament zu erfahren. Die Forscher haben drei Ziele: die Erprobung eines Evaluierungsprotokolls, die Bewertung des Wirksamkeitspotenzials und die Beurteilung der Serumspiegel aktiver Metaboliten in der Patientenpopulation.
Das Medikament wirkt im Labor, aber die Forscher wissen noch nicht, ob es bei den Patienten wirkt. Bei dieser First-in-Disease-Studie geht es wirklich um Sicherheit und Verträglichkeit, aber die Forscher werden auch prüfen, ob es einen klinischen Nutzen gibt.
Hier mehr Info (auf Englisch): Forschung mit Phenybutyrat
Wie wirkt 4-phenylbutyrat?
Phenylbutyrat ist ein Medikament, das als chemisches Chaperon wirken kann, um das Protein STXBP1 bei der Proteinfaltung zu stabilisieren.
Eine Proteinfaltung ist aufgrund von stxbp1-Mutationen in der Aminosäuresequenz bei STXBP1 Kindern nicht möglich. Proteinfaltung ist der Prozess, durch den eine Proteinstruktur ihre funktionelle Form oder Konformation annimmt.
Durch die Faltung in eine spezifische 3-dimensionale Form ist das Protein Syntaxin-1 in der Lage, seine biologische Funktion zu erfüllen, was notwendig ist, damit die Neuronen im Gehirn miteinander kommunizieren und richtig funktionieren können.
Syntaxin-1 ist Teil des SNARE-Komplexes, der die Vesikelfusion in die Neuronenzellmembran vorbereitet und die Freisetzung des Neurotransmitters entlang der Synapsen erleichtert. Eine Änderung der Neurotransmitterspiegel kann zu einer unkontrollierten Aktivierung (Erregung) von Neuronen im Gehirn führen, was zu epileptischen Anfällen und andere Störungen führt.
Syntaxin-1 ist für die Transmitterfreisetzung von der Präsynapse eines Neurons zur Postsynapse eines anderen Neurons erforderlich. (Es ist der Schlüsselmechanismus, mit dem Neuronen miteinander kommunizieren.).
(siehe auch die Bilder auf der stxbp1.de Website, button "Was ist STXBP1?")
Was ist 4-Phenylbutyrat?
4-Phenylbutyrat ist ein chemisches Chaperon, das dazu in der Lage ist:
- Proteinspiegel von Munc18-1 (stxbp1) zu stabilisieren und neuronale Defizite zu korrigieren
- die Neurotransmitter-Freisetzung zwischen den Synapsen der Neuronen zu erhöhen bzw. zu verbessern
- unsachgemäß (falsch) gefaltete Proteine zu stabilisieren bzw. zu korrigieren
- betroffene Proteine effizienter zu falten und sie zum geeigneten intrazellulären oder extrazellulären Ziel zu transportieren
Was sind Chaperone? Chaperone sind Proteine, die die neu synthetisierten Proteine bei der Faltung bzw. die neu synthetisierten Proteine "helfen", sich korrekt zu falten.
Eine erfolgreiche 4-Phenylbutyrat-Therapie könnte daher STXBP1 Kindern helfen, da ihnen das Protein Syntaxin-1 fehlt oder es fehlerhaft ist.
Das 4-Phenylbyturat könnte die Defizite bei der Neurotransmitterfreisetzung mit Munc18-1 (stxbp1) Mutationen signifikant beheben und die synaptische Funktion bei den STXBP1 Kindern wieder auf ein normales Funktionsniveau zurückbringen.
Hoffen wir auf eine Heilung für die STXBP1 Kinder. Phenylbutyrat wird nun an 10 STXBP1 Kinder getestet.
Hier geht es zum Pre–Print der Studie:
Pre–Print Ravicti