Sammlung von genetischen Infografiken
Hier entsteht eine Sammlung von genetischen Grafiken, die Genetik auf Deutsch und auf Englisch erklären sollen! Der Download dieser Infografiken ist nicht erlaubt!!!
Transposons – nichtvirale Vektoren für die Gentherapie
Transposons sind mobile DNA-Sequenzen, die eine fundamentale Rolle bei der Evolution und Strukturierung von Genomen spielen. Sie sind hocheffiziente Werkzeuge fü die Mutationsgenese, mit denen Wissenschaftler Genfunktionen entschlüsseln, Krankheitsmechanismen aufdecken und genomweite Screenings durchführen können.
In der modernen Molekularbiologie sind Transposon-Systeme, die aus mobilisierbaren DNA-Sequenzen – den "springenden Genen" – und dem entsprechenden Transposase-Enzym bestehen, von großer Bedeutung, weil sie das Hinzufügen, Entfernen oder Neuanordnen von genetischem Material ermöglichen, ohne dass wirtseigene Reparaturmechanismen genutzt werden.
In der biomedizinischen Forschung sind die großen Genomsequenzierungen, die künstliche Insertionsmutagenese zur Genfunktionsentdeckung und die Transgenese bei vielen Modelltieren die Hauptanwendungsgebiete dieser hocheffizienten genetischen Werkzeuge.
In der Neuroentwicklungsforschung, vor allem bei der Analyse des für die synaptische Neurotransmitter-Freisetzung wichtigen STXBP1-Gens, sind hochentwickelte Transposon-Plattformen wie das Sleeping Beauty- oder das piggyBac-System eine verlässliche, nicht-virale Methode, um schwere, frühkindliche Entwicklungs- und epileptische Enzephalopathien zu simulieren.
Indem sie Transposons mit großen, funktionsfähigen menschlichen STXBP1-Transgenen in Tiermodelle oder patientenbasierte Stammzellen einbringen, können Forscher schnell pathologische Mechanismen untersuchen, therapeutische Grenzen identifizieren und gezielt nach Präzisionsmedikamenten suchen.
In klinischen und experimentellen Anwendungen sind diese Transposon-Vektoren anderen Genbearbeitungswerkzeugen oft überlegen, weil sie Frachten von bis zu 100 Kilobasen stabil integrieren kann, ohne dass toxische Doppelstrangbrüche oder weitreichende DNA-Schadensreaktionen in der Zelle verursacht werden. Außerdem sind diese Systeme im Vergleich zu viralen Vektoren kostengünstiger und einfacher in großen Stückzahlen herzustellen, während sie eine geringere Immunogenität und ein gut kalkulierbares Integrationsprofil aufweisen, das man sogar mathematisch beschreiben kann.
Trotz ihrer Vorteile bringt die Anwendung der Transposon-Technologie erhebliche biologische Risiken mit sich, wie Off-Target-Effekte, unbeabsichtigte genomische Umlagerungen und die Gefahr einer unkontrollierten Insertionsmutagenese, bei der das Transgen zufällig vitale Wirtsgene beschädigt, was Onkogenese oder genomische Instabilität zur Folge haben kann.
Ein weiteres technisches Risiko ist die Überproduktionshemmung, bei der eine schlecht regulierte Expression der Transposase die Insertionseffizienz stark mindert oder sogar dazu führt, dass das Enzym das Genom nach Erreichen des ursprünglichen therapeutischen Ziels durch fortwährende Ausschneide- und Einfügevorgänge destabilisiert.


Therapies "from bench to bedside" – Immunotoxicity – Genotoxicity
Die translationalen Forschungswege vom Labor bis zum Patienten stehen vor erheblichen Herausforderungen bei der Bewertung von Immunotoxizität und Genotoxizität neuartiger Therapien.
Gentherapien mit Adeno-assoziierten Viren (AAV) bieten vielversprechende Möglichkeiten, genetische Mutationen direkt in Neuronen des Gehirns zu korrigieren. Je nach Mutation liefert das AAV-Kapsid eine gesunde Genkopie als Ersatz oder bringt molekulare Werkzeuge wie RNA-Interferenz oder CRISPR mit, um schädliche, mutierte Gene gezielt auszuschalten.
Forschende verwenden hochentwickelte Kapside wie AAV9, die die Blut-Hirn-Schranke nach Injektion in die Blutbahn effizient überwinden, oder verabreichen den Vektor lokal in die betroffene Gehirnregion.
Für eine erfolgreiche Anwendung im Gehirn müssen jedoch immunologische und genetische Sicherheitsmechanismen präzise aufeinander abgestimmt werden.
Immunotoxizität umfasst Schäden am Immunsystem oder immunbezogenen Prozessen, die durch die Therapie oder deren Metaboliten verursacht werden und langfristige klinische Folgen haben können. Sie stellt ein großes Risiko dar, da das Immunsystem des Patienten sowohl das virale Kapsid als auch das neu gebildete Transgen-Protein als fremd erkennen kann. Besitzt der Patient bereits vorbestehende neutralisierende Antikörper durch eine frühere Virusinfektion, wird der Vektor zerstört, bevor er Zielzellen erreicht. Hohe Dosen viraler Vektoren können zudem eine akute Entzündungsreaktion auslösen, die als Zytokinsturm bekannt ist.
Um unerwünschte Abwehrreaktionen zu minimieren, werden optimierte Kapside entwickelt, die das Immunsystem weniger leicht erkennen, während begleitende Medikation die Immunantwort während der Verabreichung sanft reguliert.
Genotoxizität betrifft direkte Schäden an der DNA, die Krebs verursachen können. Obwohl AAV-Vektoren weitgehend außerhalb des Genoms verbleiben, kann es selten zu ungezielter Integration kommen. Falls das Transgen in ein Tumorsuppressorgen oder in der Nähe eines Onkogens landet, kann dies die Zellteilung unkontrolliert anregen und zu Tumoren führen.
Um Genotoxizität zu beherrschen und das neuronale Erbgut zu schützen, werden therapeutische Gene mit spezifischen biologischen Schaltern ausgestattet: Promotoren sorgen dafür, dass das Gen nur in Zielneuronen aktiv wird, selbstinaktivierende Vektorsysteme verhindern zusätzlich, dass benachbarte Gene im Erbgut der Nervenzelle versehentlich aktiviert werden.
Der intravenöse Weg über die Blutbahn bringt spezifische Herausforderungen, da sich das Virus im ganzen Körper verteilt und ein Großteil der Dosis in der Leber landet. Um das Immunsystem nicht zu überlasten und Entzündungsreaktionen wie einen Zytokinsturm zu vermeiden, wird vorab auf bestehende Antikörper gegen AAV9 getestet und während der Behandlung eine vorübergehende Medikation eingesetzt.
Durch gewebespezifische Kontrollschalter, maßgeschneiderte Kapsiden und moderne Genbearbeitungswerkzeuge lässt sich die Gentherapie im Gehirn heute so steuern, dass sie therapeutisch wirkt und gleichzeitig ein hohes Maß an Sicherheit und einen sicheren Pfad für die Medizin der Zukunft bietet.


Gentests vs. Genomtests
Genetische und genomische Tests sind zu unverzichtbaren Werkzeugen in der modernen Medizin geworden und bieten Einblicke, die von der individuellen Diagnose über die Risikobewertung in der Familie bis hin zur öffentlichen Gesundheit reichen.
Genetische Tests beziehen sich typischerweise auf die Analyse eines spezifischen Gens oder einer kleinen Gen-Gruppe, um Mutationen zu identifizieren, die eine erblich bedingte Erkrankung verursachen oder dazu beitragen.
Genomische Tests decken ein breiteres Spektrum ab, indem sie große Teile des Genoms untersuchen, wie das gesamte Exom (WES) oder das gesamte Genom (WGS), um Varianten in vielen Genen zu erkennen und atypische oder unerwartete genetische Ätiologien zu identifizieren.
Die gesamte Exom- und Genom-Sequenzierung untersucht das gesamte kodierende oder nicht-kodierende Genom, um Varianten zu identifizieren, die komplexe oder undiagnostizierte Krankheiten, insbesondere seltene Erkrankungen, erklären können.
Bei seltenen Krankheiten sind genomische Ansätze oft am informativsten als erster Schritt. Typischerweise umfasst diese Reihenfolge Paneltests für vermutete Syndrome, Whole Exome Sequencing (WES) oder Whole Genome Sequencing (WGS), um den diagnostischen Ertrag zu maximieren. Trio-Tests (Proband plus beide Eltern) verbessern häufig die Interpretierbarkeit. Kaskadentests – das Testen von Familienmitgliedern, nachdem eine wahrscheinliche Diagnose gestellt wurde – können Erbmustern bestätigen und das Risiko für Verwandte klären. Kaskadentests für Familienmitglieder werfen Überlegungen zu Zufallsbefunden und der Notwendigkeit einer angemessenen Vor- und Nachtestberatung auf.
Pharmakogenomische Tests, ein zentraler Aspekt der personalisierten Medizin, untersuchen, wie genetische Variationen die Reaktion eines Individuums auf Medikamente beeinflussen. Durch die Identifizierung von Varianten, die den Arzneimittelstoffwechsel, die Wirksamkeit und das Risiko von Nebenwirkungen beeinflussen, informiert die Pharmakogenomik über die Arzneimittelauswahl und Dosierung, mit dem Ziel, den therapeutischen Nutzen zu maximieren und gleichzeitig Schäden zu minimieren.
Zusammen integrieren genetische, genomische und pharmakogenomische Tests erbliche Informationen mit klinischem Kontext, um Präventions-, Diagnose- und Behandlungsstrategien maßzuschneidern, was letztendlich die Ergebnisse verbessert und Trial-and-Error in der Patientenversorgung reduziert.
Ethische, rechtliche und soziale Implikationen, Datenschutz und gerechter Zugang sind kritische Überlegungen, da diese Technologien in der routinemäßigen Praxis immer verbreiteter werden.


STXBP1-RD – unterschiedliche genetische Ursachen
Der Ursprung und der individuelle Verlauf genetischer Erkrankungen hängen von völlig unterschiedlichen biologischen Mechanismen ab.
Bei klassisch vererbten Krankheiten wird die genetische Veränderung direkt von den Eltern an die Kinder weitergegeben, sodass die Mutation von Beginn an in jeder einzelnen Zelle des Nachkommens vorliegt. Dies ist bei STXBP1-RD jedoch äußerst selten, da Betroffene aufgrund der Schwere der Erkrankung meist keine eigenen Kinder bekommen.
Im Fall von STXBP1-RD treten De-novo-Mutationen allerdings äußerst häufig auf; Schätzungen zufolge liegt der Anteil solcher Neumutationen bei etwa 95 Prozent. Diese Veränderungen im Erbgut erscheinen erstmals im betroffenen Kind und sind bei den Eltern in den Körperzellen meist nicht nachweisbar. Solche Neumutationen entstehen spontan in einer Keimzelle der Eltern oder in den ersten Zellteilungen des Embryos. Die Eltern selbst sind asymptomatisch und vollkommen gesund, weshalb die Erkrankung ohne familiäre Vorgeschichte auftritt.
Findet eine solche Spontanmutation erst zu einem späteren Zeitpunkt der Embryonalentwicklung statt, kann beim Kind ein somatisches Mosaik entstehen, bei dem nur ein Teil der Körperzellen den Defekt trägt, während die restlichen Zellen gesund bleiben. Mosaizismus beschreibt generell das Vorhandensein zweier oder mehrerer genetisch unterschiedlicher Zelllinien in einem Individuum. Ein Mosaizismus kann jedoch auch isoliert in den Ei- oder Samenzellen eines eigentlich gesunden Elternteils vorliegen. Ein solches Keimbahnmosaik führt dazu, dass das Kind die Mutation voll vererbt bekommt und die Erkrankung in allen Zellen trägt, obwohl die Eltern keine Symptome zeigen.
Eine Wiederholung einer Keimbahn- bzw. gonadales Mosaik ist in Folgegenerationen jedoch möglich, weshalb eine genetische Beratung sinnvoll sind.
Schließlich spielen Modifikator-Gene eine bedeutsame Rolle für den klinischen Verlauf. Sie bestimmen maßgeblich, wie schwer die Erkrankung im Alltag ausgeprägt ist. Diese Gene verursachen die Krankheit zwar nicht selbst, können aber die Wirkung des Hauptgens verändern, verstärken oder dämpfen. Dies erklärt, warum selbst Menschen mit exakt derselben STXBP1-Mutation völlig unterschiedliche Krankheitsbilder und Symptome zeigen können.


Genomik vs. Genetik
Die Erforschung des Erbguts hat das Verständnis vom Leben und den biologischen Prozessen grundlegend verändert. Im Zentrum dieser Wissenschaft stehen die Genetik und die Genomik, zwei eng miteinander verwandte, aber unterschiedlich weit gefasste Disziplinen. Als Basis dient die DNA, die wie ein universelles Rezeptbuch die Bauanleitung für jeden Organismus enthält. Die klassische Genetik konzentriert sich dabei auf die Untersuchung einzelner Gene, also jener spezifischen DNA-Abschnitte. Die moderne Genomik hingegen geht einen entscheidenden Schritt weiter und betrachtet das Genom als Ganzes.
Ein Genom ist die Gesamtheit des genetischen Materials eines Organismus und umfasst sowohl alle seine Gene als auch die nichtkodierenden Sequenzen, die steuern, wann, wo und wie Gene exprimiert werden. Es enthält die Informationen, die für den Aufbau und Erhalt des Organismus während seines gesamten Lebens notwendig sind, und variiert zwischen Individuen und Populationen, wodurch es Merkmale, Gesundheit und Anfälligkeit für Krankheiten prägt.
Die Genetik konzentriert sich auf die Erforschung einzelner Gene und ihrer Rolle bei der Vererbung und Variation. Sie stellt Fragen wie: Wie beeinflusst ein einzelnes Gen ein Merkmal oder die Veranlagung für eine Krankheit, und wie werden Merkmale von einer Generation an die nächste weitergegeben?
Die Genomik hingegen untersucht das gesamte Genom und die Wechselwirkungen zwischen all seinen Teilen. Sie nutzt groß angelegte Datensätze, um komplexe biologische Systeme, Signalwege und Netzwerke zu verstehen, und erforscht, wie Kombinationen vieler Gene zusammen mit Umweltfaktoren zu Gesundheit und Krankheit beitragen.
Beide Perspektiven sind unverzichtbar. Die Genetik liefert tiefe Einblicke in bestimmte Gene und die Mendelsche Vererbung und bildet die Grundlage für gezielte Diagnosen und Behandlungen. Die Genomik bietet ein umfassenderes Verständnis auf Systemebene, das entscheidend ist, um komplexe Krankheiten zu entschlüsseln, die personalisierte Medizin voranzutreiben und Erkenntnisse auf Populationsebene zu gewinnen.
Zusammen ermöglichen die Genetik und die Genomik ein vollständigeres Bild von Biologie, Krankheitsrisiken und therapeutischen Möglichkeiten. Durch bahnbrechende Technologien in der Genomsequenzierung revolutionieren sie die moderne Medizin. Sie ermöglichen den Übergang von einer standardisierten Behandlung hin zu einer personalisierten Medizin, bei der Therapien exakt auf das genetische Profil des einzelnen Patienten abgestimmt werden.


L-Serin ist eine proteinogene, nicht essentielle Aminosäure
L-Serin ist eine proteinogene, nicht essentielle Aminosäure, die eine vielfältige Rolle im Zellstoffwechsel und in der Nervenphysiologie spielt. Im Menschen gilt sie als nicht essenziell, da der Körper sie selbst herstellen kann, wird jedoch häufig auch als Nahrungsergänzungsmittel eingenommen.
L-Serin dient als Baustein für Proteine und fungiert als Vorläufer für weitere wichtige Moleküle, darunter Glycin und Cystein. Im Zentralnervensystem fungiert L-Serin als Vorläufer von D-Serin, das durch das Enzym Serinracemase synthetisiert wird und als Co-Agonist an NMDA-Glutamatrezeptoren wirkt, wodurch es die synaptische Plastizität und die exzitatorische Neurotransmission beeinflusst.
In der Forschung wird L-Serin zur Untersuchung des Aminosäurestoffwechsels, der Neurotransmitterregulation sowie der molekularen Mechanismen eingesetzt, die der neurologischen Entwicklung und neurodegenerativen Erkrankungen zugrunde liegen. Es wird in der Forschung zu einer Vielzahl von Erkrankungen eingesetzt, da sein Stoffwechsel mehrere kritische Stoffwechselwege berührt.
Aufgrund seiner Beteiligung an mehreren miteinander verbundenen Stoffwechselwegen wird L-Serin häufig untersucht, um zu verstehen, wie Veränderungen in der Verfügbarkeit von Aminosäuren die neuronale Funktion, die metabolische Homöostase und Krankheitsprozesse beeinflussen können. Es wird auch zur Modulation verwandter Stoffwechselwege in Zellkulturen und Tiermodellen verwendet, um seine Auswirkungen auf die Proteinsynthese, Methylierungsreaktionen und die Membranbiogenese zu untersuchen. Bei neurodegenerativen und neurologischen Entwicklungsstörungen haben Forscher in einigen Kontexten die Serin-Supplementierung als therapeutischen Ansatz untersucht.
Was den Stoffwechsel betrifft, so wird L-Serin aus der Nahrung oder aus Nahrungsergänzungsmitteln im Dünndarm resorbiert. Je nach zellulärem Bedarf kann es auch in andere Stoffwechselwege einfließen, einschließlich der Energieproduktion.
Nebenwirkungen oder unerwünschte Wechselwirkungen mit Medikamenten sind möglich und können je nach spezifischem Medikament und individuellen Faktoren variieren. Zu den potenziellen Bedenken zählen Wechselwirkungen mit Medikamenten, die den Aminosäuretransport oder -stoffwechsel beeinflussen, und eine hochdosierte Supplementierung kann die Folat- oder Glycin-Stoffwechselwege in einer Weise beeinflussen, die zu Wechselwirkungen mit bestimmten Therapien führen könnte.
Wie bei jedem Nahrungsergänzungsmittel ist es wichtig, vor der Kombination von L-Serin mit verschreibungspflichtigen Medikamenten, insbesondere im Zusammenhang mit neurologischen, metabolischen oder Krebstherapien, medizinisches Fachpersonal zu konsultieren, um Sicherheit, Dosierung und mögliche Wechselwirkungen beurteilen zu können.


Medikamente – Nebenwirkungen vs. unerwünschte Wirkungen
Bei der Erörterung von Medikamenten ist es wichtig, zwischen Nebenwirkungen und unerwünschten Wirkungen zu unterscheiden. Diese Begriffe beschreiben unterschiedliche Erfahrungen, die Patienten nach der Einnahme eines Medikaments machen können, und das Verständnis dieses Unterschieds hilft sowohl Patienten als auch medizinischem Fachpersonal, die Therapie sicherer und effektiver zu gestalten.
Nebenwirkungen sind unbeabsichtigte, aber vorhersehbare Effekte, die bei normalen therapeutischen Dosen auftreten und oft gut verträglich oder beherrschbar sind. Unerwünschte Wirkungen hingegen sind schädliche, unerwartete oder unerträgliche Effekte, die möglicherweise das Absetzen des Medikaments oder die Inanspruchnahme dringender medizinischer Hilfe erfordern.
Nebenwirkungen sind häufig und werden von Ärzten bei der Verschreibung oft erwartet. Sie können von leicht (Übelkeit, Mundtrockenheit) bis hin zu störender, aber nicht lebensbedrohlich (Müdigkeit, Schwindel) reichen. Unerwünschte Wirkungen sind schwerwiegender, potenziell gefährlich oder können in einigen Fällen lebensbedrohlich sein, wie z. B. schwere Leberschäden oder schwere Hautreaktionen.
Nebenwirkungen sind in der Regel dosisabhängig und zu erwarten, während unerwünschte Wirkungen weniger vorhersehbar sein können und dringende Maßnahmen erfordern.
Das Wissen darüber, ob es sich bei einer Wirkung um eine häufige Nebenwirkung oder eine unerwünschte Wirkung handelt, leitet klinische Entscheidungen. Bei Nebenwirkungen können Ärzte Strategien zu deren Bewältigung anbieten, die Dosierung anpassen oder auf Alternativen umstellen, während die Therapie fortgesetzt wird. Bei unerwünschten Wirkungen steht die Patientensicherheit im Vordergrund: Absetzen des betreffenden Medikaments, rasche Beurteilung und Suche nach sichereren Alternativen.
Diese Unterscheidung unterstützt auch die personalisierte Medizin, da bei manchen Personen je nach genetischer Veranlagung, Allgemeinzustand und gleichzeitig eingenommenen Medikamenten unterschiedliche Muster von Nebenwirkungen oder unerwünschten Wirkungen auftreten können.
Die Prävention oder Risikominderung beginnt mit einer sorgfältigen Verschreibung und Patientenaufklärung, einschließlich der Erörterung bekannter Nebenwirkungen und Warnzeichen für unerwünschte Wirkungen vor Beginn der Therapie. Regelmäßige Überwachung und Nachsorge ermöglichen die frühzeitige Erkennung von unerträglichen Wirkungen oder gefährlichen Reaktionen.
Pharmakogenetische Tests bieten eine Möglichkeit, die Wahl der Medikamente auf das genetische Profil einer Person abzustimmen.
Pharmakogenetische Erkenntnisse können als Leitfaden für die Dosierung oder die Auswahl alternativer Therapien dienen, um Reaktionen zu vermeiden, die mit Stoffwechselenzymen oder Arzneimitteltransportern zusammenhängen.
Die Pharmakogenetik ist jedoch keine Garantie, aber pharmakogenetische Tests können die Wahrscheinlichkeit schädlicher Reaktionen verringern und die Behandlungsergebnisse insgesamt verbessern.


CRISPr-Cas9 vs. CRISPRa – CRISPr-Systeme
Die Entwicklung der CRISPr-Technologie (Clustered Regularly Interspaced Short Palindromic Repeats) hat eine neue Ära in der biomedizinischen Forschung eingeleitet und die Fähigkeit zur Genomeditierung grundlegend verändert.
CRISPr-Cas9 ist ein molekulares Werkzeug. Es erlaubt die DNA gezielt zu schneiden und damit Gene zu verändern. Cas9 ist ein Enzym, das an einer vorbereiteten RNA-Vorlage bindet und die DNA an einer festgelegten Stelle schneidet.
Ursprünglich als bakterieller Abwehrmechanismus gegen Viren entdeckt, wurde CRISPR-Cas9 im Jahr 2012 von Doudna und Charpentier als effizientes Werkzeug für die Genbearbeitung modifiziert.
Seitdem wird CRISPr-Cas9 in Forschung genutzt, um Gene zu deaktivieren, zu korrigieren oder neue Funktionen einzuführen. Das Cas9-Enzym erzeugt gezielte Doppelstrangbrüche, was das präzise Einfügen, Löschen oder Verändern von genetischem Material ermöglicht.
Um diese Plattform zu erweitern, wurde die CRISPR-Aktivierung (CRISPRa) entwickelt. CRISPRa erlaubt anstelle eines Schnitts eine transkriptionelle Steuerung. Dieser Ansatz nutzt eine katalytisch inaktive Cas9 (dCas9) in Kombination mit Aktivatoren, um die Expression spezifischer Gene hochzuregulieren.
Diese Werkzeuge bieten ein enormes Potenzial für die Behandlung neurobiologischer Erkrankungen wie den STXBP1-assoziierten Enzephalopathien, bei denen insbesondere CRISPRa eingesetzt werden kann, um die Aktivität der verbleibenden gesunden Genkopie des Patienten zu verstärken und so die Haploinsuffizienz therapeutisch auszugleichen.


Aggressives Verhalten – Selbstverletzung – Lebensqualität?
Zusätzlich zu den bestehenden Problemen verändert sich im Alltag und in der Lebensqualität der Familien mit einem behinderten Kind, insbesondere bei einem nonverbalen, plötzlich vieles. Es geht nicht mehr ausschließlich um die Epilepsiebehandlung mit Antiepileptika; es rücken vermehrt auch psychopharmakologische Therapien in den Fokus.
Das Kind zeigt plötzlich Verhaltensänderungen, für die den Eltern oft kein eindeutiger Auslöser erkennbar ist. Es kommt zu Schlagen, Beißen, Verspritzen von Speichel, Werfen von Gegenständen sowie zu starken Unruhe- und Aggressionsreaktionen.
Oft entstehen daraus blaue Flecken und Verletzungen an Stirn, Wangen, Armen und Hüften. Diese Verhaltensweisen beeinträchtigen den Alltag der Familie massiv und führen dazu, dass gemeinsame Aktivitäten mit dem verletzten Kind wie Einkäufe oder Geburtstagsfeiern in Frage gestellt werden.
Psychopharmaka? Es werden verschiedene Medikamente ausprobiert. Einige scheinen die Situation zu verschlechtern, andere helfen nicht. Es stellt sich die Frage, ob Dosis, Verträglichkeit oder Wechselwirkungen mit den Antiepileptika eine Rolle spielen. Eltern geraten zunehmend in Ratlosigkeit.
Ein pharmakogenetischer Test kann möglicherweise helfen, die passenden Medikamente anhand des genetischen Profils des Kindes zu identifizieren.
"Personalised Medicine" zielt darauf ab, Nebenwirkungen und Wechselwirkungen zu minimieren und die oft langwierige "Trial-and-Error"-Suche nach geeigneten Therapien zu beenden.
Die Kenntnis pharmakogenetischer Zusammenhänge ermöglicht den Übergang von einer empirischen Behandlung hin zu einer personalisierten Therapie, die Wirksamkeit maximiert und toxische Effekte reduziert.


Pharmakogenetik – Zusammenspiel Gene und Enzyme?
Die Pharmakogenetik stellt ein zukunftsweisendes Bindeglied zwischen der Genetik und der klinischen Pharmakologie dar, indem sie untersucht, wie die individuelle genetische Ausstattung die Reaktion auf Arzneimittel beeinflusst. Besonders in den Fachbereichen der Neurologie und Psychiatrie, in denen die Behandlung von Epilepsie und psychischen Erkrankungen oft durch eine hohe Variabilität im Therapieansprechen sowie durch häufige Nebenwirkungen geprägt ist, gewinnt dieser Ansatz zunehmend an Bedeutung. Die Pharmakogenetik untersucht, wie genetische Unterschiede die Wirkung und den Metabolismus von Medikamenten beeinflussen.
Bei Epilepsie und psychischen Erkrankungen spielen Enzyme eine zentrale Rolle bei der Umwandlung und dem Abbau von Arzneistoffen. Variationen in Genen, die diese Enzyme codieren, führen zu individuellen Unterschieden in der Plasmaspitzenkonzentration, der Wirkdauer und dem Nebenwirkungsprofil von Therapien. Erkenntnisse aus der Pharmakogenetik ermöglichen es, Vorhersagen darüber zu treffen, wie Patienten auf bestimmte Antiepileptika oder Psychopharmaka reagieren, welche Dosierungen wahrscheinlich sicher und wirksam sind und welche Risiken für Nebenwirkungen bestehen.
Zu den Schlüsselgenen gehören spezifische Gene und die von ihnen kodierten Enzyme, die den Metabolismus von Antiepileptika und Psychopharmaka steuern. Während Variationen in den Genen des Cytochrom-P450-Systems maßgeblich bestimmen, wie schnell oder langsam ein Wirkstoff im Körper abgebaut wird und somit die notwendige Dosierung beeinflussen, spielen immunologische Marker wie das HLA-System eine entscheidende Rolle bei der Vorhersage schwerer Überempfindlichkeitsreaktionen.
Die Kenntnis dieser pharmakogenetischen Zusammenhänge ermöglicht den Übergang von einer empirischen "Trial-and-Error"-Medizin hin zu einer personalisierten Therapieform, welche die Wirksamkeit maximiert und das Risiko für toxische Effekte minimiert.


Spliceopathien?
Spliceopathien sind Erkrankungen, die durch Defekte beim RNA-Spleißen verursacht werden – jenem zellulären Prozess, bei dem die Prä-mRNA bearbeitet wird, um reife Boten-RNA und letztlich funktionsfähige Proteine zu bilden.
Wenn das Spleißen gestört ist, kann dies zu abnormalen oder verminderten Konzentrationen essenzieller Proteine führen oder Proteine mit veränderter Struktur und Funktion hervorbringen. Diese molekularen Fehler können sich auf mehrere biologische Systeme auswirken und zu einem breiten Spektrum klinischer Erscheinungsbilder führen.
Spliceopathien manifestieren sich häufig im Bereich der Entwicklung und der Neurologie, mit geistiger Behinderung, Entwicklungsverzögerung, Krampfanfällen und autismusähnlichen Merkmalen, sowie im Bereich der Muskulatur und des Stoffwechsels, wie Myopathien und metabolischen Krisen. Das Herz-Kreislauf-System kann betroffen sein, was sich manchmal in angeborenen Herzfehlern oder Kardiomyopathie äußert, und einige Erkrankungen sind multisystemisch und gehen mit Wachstumsstörungen und Organfehlbildungen einher.
In bestimmten Kontexten kann ein fehlerhaftes Spleißen von Tumorsuppressoren oder Onkogenen zu einem erhöhten Krebsrisiko beitragen.
Zu den zugrunde liegenden Ursachen zählen häufig Mutationen an Spleißstellen, Verzweigungspunkten oder regulatorischen Elementen, die die Exon-Erkennung steuern, sowie Veränderungen an RNA-bindenden Proteinen, die die Spleißosomenaktivität regulieren, und epigenetische oder transkriptionelle Faktoren, die die Spleißdynamik beeinflussen.
Die Diagnose stützt sich in der Regel auf Gentests und RNA-Sequenzierung zur Identifizierung abnormaler Transkripte, ergänzt durch Funktionsstudien an patienteneigenen Zellen. Neue Therapien wie Antisense-Oligonukleotide zur Umleitung des Spleißens, kleine Moleküle, die Spleißfaktoren modulieren, und in einigen Fällen Gentherapie sind vielversprechend für eine gezielte Behandlung.


Klinische Studien mit oder ohne Placebo?
Klinische Studien dienen dazu, festzustellen, ob eine neue Behandlung einen echten Nutzen gegenüber der Standardversorgung oder keiner Behandlung bietet. Studien mit Placebo-Kontrolle vergleichen eine experimentelle Intervention mit einer simulierten oder wirkungslosen Behandlung, um den tatsächlichen therapeutischen Effekt von Erwartungen, dem natürlichen Krankheitsverlauf oder statistischen Schwankungen zu trennen. Dieses placebokontrollierte Studiendesign tr%auml;gt zur Gewährleistung der internen Validität bei, indem es Verzerrungen reduziert und eine klarere Bewertung von Wirksamkeits- und Sicherheitssignalen ermöglicht.
Im Gegensatz dazu konzentrieren sich Studien, die ohne Placebo durchgeführt werden und häufig einen aktiven Komparator oder einen Standardversorgungsarm verwenden, auf den Vergleich der neuen Intervention mit bestehenden Therapien oder Verfahren. Diese Nicht-Placebo-Designs können die ethische Akzeptanz erhöhen, insbesondere wenn ein Behandlungsverzicht unangemessen ist oder wenn objektive Ergebnisse leicht beobachtbar sind; sie können jedoch Herausforderungen bei der Unterscheidung der tatsächlichen Wirkung der neuen Therapie von Störfaktoren mit sich bringen.
Bei seltenen Krankheiten kommt der Wahl zwischen Placebo-kontrollierten und Nicht-Placebo-Designs zusätzliche Bedeutung zu. Kleine Patientengruppen schränken die statistische Aussagekraft zur Erkennung signifikanter Unterschiede ein, sodass die Maximierung der Informationen pro Teilnehmer entscheidend wird.
Placebo-kontrollierte Studien können strenge Wirksamkeitsnachweise liefern, können jedoch ethische Bedenken aufwerfen, wenn wirksame Therapien existieren oder wenn Teilnehmer im Placebo-Arm einem erheblichen Risiko ausgesetzt sind.
In solchen Kontexten können adaptive oder angereicherte Studienkonzepte, die Verwendung historischer Kontrollen oder Studien mit aktiven Vergleichspräparaten in Betracht gezogen werden, um wissenschaftliche Validität mit Patientensicherheit und Durchführbarkeit in Einklang zu bringen.
Unabhängig vom Studienkonzept sind Transparenz bei der Definition der Endpunkte, eine sorgfältige Berücksichtigung des natürlichen Krankheitsverlaufs und eine robuste Sicherheitsüberwachung unerlässlich, um zuverlässige Evidenz zu generieren, die regulatorische Entscheidungen und letztlich den Zugang zu Therapien für Menschen mit seltenen Krankheiten unterstützen kann.


Neuromodulation für Epilepsie
Die Neuromodulation ist eine interessante Behandlungsmethode für bestimmte Formen der Epilepsie, da sie die Häufigkeit und Schwere von Anfällen ohne chirurgischen Eingriff verringert. Sie kann zum Einsatz kommen, wenn Medikamente allein nicht ausreichend wirken, und stellt möglicherweise eine Option für Menschen dar, die keine geeigneten Kandidaten für eine Operation zur Entfernung von Hirngewebe sind.
Diese Behandlungen können dazu beitragen, die Bereiche des Gehirns zu stabilisieren, die während Anfällen nicht richtig funktionieren, indem sie die Kommunikation zwischen den Nervenzellen verändern, anstatt Gewebe zu entfernen. Sie können auch die Lebensqualität der Betroffenen verbessern, wobei die Risiken und Vorteile unterschiedlich sind.
Zu den verschiedenen Optionen gehören eine Reihe von implantierbaren und schmerzfreien Methoden. Bei der Vagusnervstimulation (VNS) wird der linke Vagusnerv regelmäßig mit Stromimpulsen stimuliert, die von einem in die Brust implantierten Impulsgenerator ausgehen. Nach einigen Monaten der Therapie kann dies bei manchen Menschen zu einer Verringerung der Anfälle führen.
Die reaktive Neurostimulation (RNS) zielt direkt auf die Anfallaktivität ab, indem sie über implantierte Elektroden, die mit einem Neurostimulator verbunden sind, elektrische Stimulation an bestimmte Anfallherde im Gehirn sendet.
Die transkranielle Magnetstimulation (TMS) und die transkranielle Gleichstromstimulation (tDCS) sind nichtinvasive Verfahren, die die Erregbarkeit der Großhirnrinde verändern. Sie haben in einigen Studien leichte Wirkungen gezeigt und werden in der Regel nicht allein, sondern in Kombination mit anderen Behandlungen eingesetzt.
Die Neuromodulation hat für verschiedene Menschen unterschiedliche Vorteile, aber in einigen Fällen kann sie die Anzahl der Anfälle verringern, die mit Anfällen verbundenen Symptome lindern, die Wachsamkeit steigern, die Lebensqualität verbessern und sogar die Abhängigkeit von Anfallsmedikamenten verringern.
Epilepsiezentren mit multidisziplinären Teams aus Epileptologen, Neurochirurgen, Neuroradiologen, Neuropsychologen und spezialisierten Gerätetechnikern sind die besten Anlaufstellen für diese Art von Behandlungen. Die Beratung vor der Operation und die Nachsorge nach dem Eingriff sind sehr wichtig, um die besten Ergebnisse zu erzielen und eventuell auftretende Probleme zu bewältigen.
Epilepsie verstehen: Neuromodulation –
Fortbildungsreihe der Oskar Killinger Stiftung


? X- und Y-Chromosomen – ein wichtiges Paar ?
Die X- und Y-Chromosomen bestimmen das biologische Geschlecht und beeinflussen auf unterschiedliche Weise Vererbungsmuster, den Gengehalt und das Krankheitsrisiko.
Die X- und Y-Chromosomen stammen aus derselben Vorläuferquelle und entstanden durch einen Prozess, der als Geschlechtschromosomendifferenzierung bezeichnet wird. Zu Beginn der Evolution der Wirbeltiere gab es zwei ähnliche Geschlechtschromosomen (X und Y), die sich über Millionen von Jahren hinweg auseinanderentwickelten.
Das menschliche X-Chromosom ist groß und enthält viele Gene; das Y-Chromosom ist kleiner und trägt weniger Gene, enthält jedoch Schlüsselgene für die männliche Entwicklung.
Beim Menschen haben Frauen zwei X-Chromosomen (XX) und Männer ein X- und ein Y-Chromosom (XY).
Die meisten Gene auf dem X-Chromosom werden bei Frauen von beiden X-Kopien exprimiert, aber ein X-Chromosom in jeder Zelle wird durch die X-Chromosomen-Inaktivierung deaktiviert, um die Gendosis zwischen XX-Frauen und XY-Männern auszugleichen.
Das Y-Chromosom trägt Gene, die in erster Linie an der männlichen Geschlechtsbestimmung und der Spermatogenese beteiligt sind.
Mutationen auf dem X- oder Y-Chromosom können zu X-chromosomalen bzw. Y-chromosomalen Erkrankungen führen. X-chromosomale Erkrankungen werden durch Mutationen in Genen auf dem X-Chromosom verursacht. Da Männer nur ein X-Chromosom haben, führt eine einzige pathogene Variante oft zu einer Erkrankung bei betroffenen Männern, während Frauen Trägerinnen sein können oder manchmal betroffen sind, wenn sie eine verzerrte X-Inaktivierung oder biallelische Varianten aufweisen. Beispiele für X-chromosomale Erkrankungen sind Hämophilie A und B, Duchenne- und Becker-Muskeldystrophie sowie das Rett-Syndrom.
Y-chromosomale Erkrankungen sind seltener und betreffen typischerweise die männliche Fortpflanzungsentwicklung oder Fruchtbarkeit; sie können durch Mutationen in Genen auf dem Y-Chromosom verursacht werden, beispielsweise solchen, die die Spermatogenese beeinflussen, und werden nur vom Vater auf den Sohn übertragen.
Die Vererbungsmuster sind also nicht gleich. X-chromosomal-rezessive Erkrankungen betreffen häufiger Männer; weibliche Trägerinnen sind möglicherweise nicht betroffen oder zeigen nur leichte Symptome. X-chromosomal-dominante Erkrankungen können beide Geschlechter betreffen, sind bei Männern jedoch oft schwerer ausgeprägt. Y-chromosomale Erkrankungen werden nur vom Vater auf den Sohn übertragen und betreffen ausschließlich Männer.
Der Mensch hat 22 Paare autosomaler Chromosomen und ein Paar Geschlechtschromosomen (insgesamt 46 Chromosomen).


Eye-Gaze Technologie und CVI
Die Eye-Gazing-Technologie bietet einen Einblick in die Welt nonverbaler Kinder, darunter auch solche mit einer STXBP1-assoziierten neurologischen Entwicklungsstörung und einer zerebralen Sehbehinderung (CVI). Für diese Kinder können traditionelle Kommunikationsformen eingeschränkt sein, doch der Blickkontakt bietet eine Möglichkeit, Bedürfnisse, Vorlieben und Verständnis auszudrücken.
Indem sie verfolgen, wohin ein Kind schaut, wann es schaut und wie lange es den Blick fixiert, können Betreuer und Fachkräfte Einblicke in Aufmerksamkeit, Interesse und Absichten gewinnen.
Die Eye-Gazing-Technologie kann die frühe Kommunikationsentwicklung, sowie die Teilnahme an Lernaktivitäten fördern und als Leitfaden für personalisierte Therapien und unterstützende Kommunikationsstrategien dienen.
Eye-Gaze-Technologie (Augensteuerung) bietet Kindern mit Cortical Visual Impairment (CVI) – einer neurologisch bedingten Sehbeeinträchtigung – enorme Chancen zur Kommunikation und Teilhabe. Da bei CVI nicht die Augen, sondern die Verarbeitung im Gehirn beeinträchtigt ist, muss die Technologie speziell angepasst werden, um visuelle Überforderung zu vermeiden. Bei durchdachtem Einsatz können Eye-Gazing-Tools dazu beitragen, die Teilhabe eines Kindes an Alltagsroutinen, sozialen Interaktionen und Bildungserfahrungen zu fördern, wobei Komfort, Sicherheit und die einzigartigen Fähigkeiten des Kindes im Vordergrund stehen.
Es ist wichtig, mit Spezialisten zusammenzuarbeiten, die sowohl STXBP1-RD als auch CVI verstehen, um den Ansatz auf die Stärken und Bedürfnisse jedes einzelnen Kindes abzustimmen.


Stammzellentherapie - regenerative Medizin
In der regenerativen Medizin gewinnen Stammzelltherapien zunehmend an Bedeutung, weil sie das transformative Potenzial besitzen, geschädigte Gewebe zu ersetzen, zu reparieren oder funktionell wiederherzustellen.
Stammzellen zeichnen sich durch ihre Fähigkeit aus, sich zu unbegrenzter Anzahl zu erneuern und sich in verschiedene spezialisierte Zelltypen zu differenzieren. Dadurch eröffnen sie neue Perspektiven für die Behandlung von Erkrankungen und Verletzungen, bei denen herkömmliche Therapien oft nur symptomatisch wirken oder Gewebe zunächst schwächer machen.
Von der regenerativen Heilung von Herzgewebe nach einem Herzinfarkt über die Wiederherstellung neuronaler Funktionen bei schweren Nervenschäden bis hin zur Reparatur von Gelenkknorpel und Haut können Stammzellansätze das Potenzial bieten, die Lebensqualität Betroffener deutlich zu verbessern.
Gleichzeitig stellen sich in diesem Feld komplexe wissenschaftliche, ethische und regulatorische Fragestellungen, die sorgfältig abgewogen werden müssen, um sichere, wirksame und gerechte Therapien zu entwickeln.
Bei seltenen genetischen Erkrankungen, die auf einer Mutation basieren, werden häufig körpereigene Stammzellen (autolog) entnommen, genetisch korrigiert und zurückgegeben. Die Stammzelltherapie zielt darauf ab, fehlende oder defekte Zellen durch gesunde Stammzellen zu ersetzen oder deren Funktion zu unterstützen.
Besonders vielversprechend ist die Kombination von Stammzellen mit Geneditierungs-Werkzeugen wie CRISPR-Cas9. Damit können Mutationen direkt in den Zellen korrigiert werden, bevor sie transplantiert werden. Die Wirksamkeit hängt stark von der spezifischen Erkrankung, dem Behandlungskonzept und dem individuellen Krankheitsverlauf ab, während Sicherheitsaspekte wie Infektionsrisiko, Abstoßung und mögliche Nebenwirkungen sorgfältig überwacht werden. Forschung und klinische Studien befinden sich in verschiedenen Stadien, um Wirksamkeit, Sicherheit und Langzeitfolgen besser zu verstehen.


Telomere vs. Zentromere
Telomere und Zentromere sind wesentliche Strukturen innerhalb eukaryotischer Chromosomen, die eine entscheidende Rolle bei der Aufrechterhaltung der genetischen Stabilität und der ordnungsgem&aum;ßen Zellteilung spielen.
Telomere sind sich wiederholende Nukleotidsequenzen an den Enden der Chromosomen, die als Schutzkappen fungieren und den Verlust wichtiger genetischer Informationen während der DNA-Replikation verhindern. Sie verkürzen sich mit jeder Zellteilung – ein Prozess, der mit dem Altern und der zellulären Seneszenz in Verbindung steht – und werden in bestimmten Zelltypen und unter spezifischen Bedingungen durch spezialisierte Enzyme verlängert.
Zentromere hingegen sind verengte Regionen, die als Anlagerungsstellen für den Kinetochorkomplex dienen und durch die Vermittlung der Anlagerung von Spindelfasern an Schwesterchromatiden eine korrekte Chromosomensegregation während der Mitose und Meiose gewährleisten.
Gemeinsam tragen Telomere und Zentromere zur genomischen Integrität bei, indem sie die Chromosomenenden schützen und die korrekte Verteilung des genetischen Materials an die Tochterzellen steuern.
Wenn Telomere und Zentromere nicht richtig gebildet werden, kann dies schwerwiegende zelluläre und organismische Folgen haben.
Eine zu frühe oder unkontrollierte Telomerverkürzung kann zu erhöhter Chromosomeninstabilität und vorzeitiger Zellalterung führen. Fehlbildete Telomere erhöhen das Risiko von Chromosomenabbrüchen, Rearrangierungen oder Fusionen, was genetische Instabilität und Fehlfunktionen der Zellteilung verursachen kann.
Eine fehlerhafte Zentromerstruktur kann dazu führen, dass Chromosomen nicht ordnungsgemäß getrennt werden, was zu Aneuploidie (falsche Anzahl von Chromosomen) und genetischen Erkrankungen führt.
Insgesamt kann eine gestörte Telomer- oder Zentromerbildung zu Entwicklungsstörungen, Krebsrisiken, Neurodegeneration und verminderten Heilungsprozessen beitragen. In vielen Fällen entstehen solche Probleme durch Mutationen in Genen, die an der Replikation, dem Schutz der Chromosomenenden oder der Chromosomenorganisation beteiligt sind.


Spiegelneuronen
Spiegelneuronen sind eine Klasse von Neuronen, die sowohl feuern, wenn eine Person eine zielgerichtete Handlung ausführt, als auch wenn sie eine Handlung beobachtet, die von jemand anderem ausgeführt wird.
Diese Neuronen wurden in den 1990er Jahren im prämotorischen Kortex von Makaken entdeckt und enthüllten einen neuronalen Mechanismus, der möglicherweise der Imitation, dem Verständnis von Handlungen und der Fähigkeit, die Absichten anderer zu erschließen, zugrunde liegt.
Spiegelneuronen wurden in mehreren Hirnarealen von Primaten identifiziert, darunter im prämotorischen Kortex (insbesondere im ventralen prämotorischen Areal, F5, bei Affen) und im unteren Parietallappen.
Beim Menschen wurden Hinweise auf spiegelähnliche Systeme in Regionen wie dem prämotorischen Kortex, dem inferioren Frontalgyrus (oft mit dem Broca-Areal verbunden) und Teilen des inferioren Parietallappens gefunden, wobei funktionelle Bildgebung auf ein Netzwerk hindeutet, das die Beobachtung und Ausführung von Handlungen sowie die soziale Kognition unterstützt.
Die Kernidee besteht darin, dass Spiegelneuronen eine Brücke zwischen Wahrnehmung und Handlung schlagen. Indem sie sowohl während der Ausführung als auch während der Beobachtung von Handlungen aktiviert werden, können sie dem Individuum helfen, neue Handlungen zu imitieren und zu lernen, die Handlungen und Absichten anderer zu verstehen, beobachtete Handlungen auf das eigene motorische System zu übertragen, soziales und kommunikatives Verhalten zu unterstützen und zu übergeordneten Prozessen wie Empathie und Theory of Mind beizutragen.
Die Spiegelneuronen-Hypothese hat zu umfangreichen Forschungen geführt, die ihre Rolle bei Imitation, Sprachentwicklung, Empathie und sozialen Störungen untersuchen. Sie ist jedoch auch Gegenstand von Debatten. Einige Studien stützen eine umfassende Rolle von Spiegelneuronen in der sozialen Kognition, während andere darauf hindeuten, dass die Spiegelaktivität indirekt, kontextabhängig oder weniger zentral sein könnte als ursprünglich angenommen.
Laufende Forschungen nutzen eine Kombination aus Elektrophysiologie, Neuroimaging, Neuropsychologie und Computermodellen, um das Verständnis darüber zu verfeinern, wo spiegelähnliche Aktivitäten auftreten, wie sie sich entwickeln und welche Funktionen sie unterstützen.


Cure vs. Treatment
Der Unterschied zwischen Cure (Heilung) und Treatment (Behandlung) bzw. zwischen Heilung und Behandlung ist für Eltern mit einem behinderten Kind oft von zentraler Bedeutung.
Eine Heilung bedeutet, dass die zugrundeliegende Ursache dauerhaft beseitigt wird und das Kind dauerhaft gesund ist bzw. die Erkrankung vollständig verschwindet. Eine Behandlung hingegen zielt darauf ab, Symptome zu lindern, das Fortschreiten der Erkrankung zu verlangsamen oder Komplikationen zu verhindern, ohne die Grundursache zwangsläufig zu eliminieren.
Heilung kann ein langfristiges, oft unsicheres Ziel sein, besonders bei seltenen oder Chromosomen- oder genetisch bedingten Erkrankungen. Behandlungen können dagegen meist sofortige oder baldige Verbesserungen bringen, erfordern aber oft eine kontinuierliche oder lebenslange Anwendung.
Eine Heilung verspricht dauerhaftes Wegfallen der Erkrankung, was immense Auswirkungen auf Alltag, Schule, Familie und Zukunftsperspektiven haben kann. Eine Behandlung kann die Lebensqualität deutlich erhöhen, Schmerzen lindern, Mobilität verbessern oder Kommunikationsfähigkeiten unterstützen, auch wenn die Grundursache bestehen bleibt.
Behandlungen bringen oft Nebenwirkungen, Belastungen durch regelmäßige Therapiesitzungen, Medikamentenwechselwirkungen und Kosten mit sich. Die Entscheidung für eine Behandlung bedeutet auch, diese Risiken aktiv abzuwägen und zu prüfen, ob der potenzielle Nutzen die Belastungen rechtfertigt. Bei einer Heilung entfallen oft langfristige therapeutische Belastungen, sofern sie tatsächlich dauerhaft erreichbar ist; jedoch bleiben auch in der Hoffnung auf Heilung Unsicherheiten und Risiken bestehen.
Der Zugang zu Therapien, Kliniken, spezialisierten Zentren und Kosten kann stark variieren. Heilungschancen hängen oft von Forschungsstand, Genetik und individuellen Faktoren ab und sind nicht immer gleich für alle Betroffenen. Behandlungen können dagegen häufiger verfügbar sein, erfordern jedoch oft Geduld, Budgetplanung und organisatorische Anstrengungen der Familie.
Eltern stehen vor ethischen Entscheidungen, etwa ob invasive oder experimentelle Therapien sinnvoll sind, welches Risiko akzeptabel ist und wie man Druck vermeidet, auf Heilung statt auf beständige Lebensqualität zu setzen.
Der Forschungsstand ändert sich kontinuierlich. Was heute als Behandlung gilt, kann morgen weiterentwickelt werden, und neue Therapien könnten Heilungschancen eröffnen.
Neben medizinischen Aspekten spielen auch psychologische und soziale Faktoren eine Rolle. Der Umgang mit einer Behinderung im Familienalltag, schulische Integration, Unterstützung durch Therapien, Hilfsmittel und das Erkennen von Ressourcen in der Gemeinschaft sind entscheidend. Klarheit über Ziele von Heilung oder Behandlung kann helfen, konkrete Unterstützungsbedarf zu definieren und Stress zu reduzieren.
Eltern sollten sich darüber im Klaren sein, welches Ziel realistisch erreichbar ist und welche Lebensqualität damit verbunden ist. Für Eltern bedeutet dies eine gründliche Abwägung von Realisierbarkeit, Nutzen, Belastungen, Kosten, Lebensqualitä und Zukunftsperspektivendes Kindes und der Famile.


Vom Molekül zum Drug
Drug Repurposing gewinnt zunehmend an Bedeutung, insbesondere bei seltenen Erkrankungen, weil die Entwicklung eines neuen Medikaments oft viele Jahre in Anspruch nimmt. Durch das Umwidmen eines bereits zugelassenen Medikaments lassen sich Entwicklungszeiten erheblich verkürzen, da bereits umfangreiche Daten zu Sicherheit, Verträglichkeit und Wirksamkeit vorliegen. Der zentrale Gedanke besteht darin, bestehende Substanzen für Indikationen zu nutzen, für die bislang kein geeignetes Therapiekonzept vorhanden ist. So können frühere Investitionen in Forschung und Entwicklung erneut genutzt werden, um patientenorientierte Lösungen schneller auf den Markt zu bringen.
Der Weg von einem Molekül zu einem zugelassenen Medikament umfasst mehrere gut definierte Phasen. Zunächst erfolgt eine Hypothesengetriebene Identifikation geeigneter Kandidaten, bei der Wirkmechanismen, Zielproteine und vorhandene klinische Daten systematisch bewertet werden. Danach folgt eine sorgfältige Literaturrecherche sowie eine Prüfung relevanter pharmakologischer Eigenschaften, wie Pharmakokinetik, Pharmakodynamik, Spezifität und off-target Wirkungen. Wichtige Schritte sind hier die Bewertung der Dosis-Wirkungs-Beziehung, das Sicherheitsprofil sowie potenzielle Wechselwirkungen mit anderen Therapien.
Im Weiteren stehen präklinische Studien im Fokus, die die Relevanz der neuen Indikation in passenden Modellen verifizieren. Parallel oder zeitlich danach erfolgt die Planung und Durchführung von klinischen Studien, die speziell darauf abzielen, die Wirksamkeit und Sicherheit der Reposition in der neuen Erkrankung nachzuweisen. Typischerweise beginnt dies mit Off-Label- und Phase-II-Studien, gefolgt von kontrollierten Phase-III-Studien, sofern frühere Ergebnisse vielversprechend sind. Zentral ist dabei die Berücksichtigung von regulatorischen Anforderungen: Oftmals werden Daten für eine ergänzende Zulassung oder eine neue Indikation nachgereicht, inklusive spezifischer Sicherheitsüberwachungs- und Pharmakovigilanzmaßnahmen.
Ein weiterer wichtiger Aspekt ist die Datengrundlage zur Sicherheit, da ein in einer anderen Indikation zugelassenes Medikament möglicherweise andere Nebenwirkungen oder Risikoprofile in der neuen Patientengruppe entfaltet. Daher müssen patientenspezifische Faktoren wie Alter, Komorbiditäten, Begleitmedikation und genetische Unterschiede sorgfältig berücksichtigt werden. Zudem spielen wirtschaftliche Überlegungen eine Rolle: Die Rekreation eines Medikaments kann die Kosten senken und den Zugang erleichtern, doch stets gilt es, ein tragfähiges Nutzungsprofil zu gewährleisten, das Regulatory- und Gesundheitsökonomie-Anforderungen erfüllt.
Zusammenfassend lässt sich sagen, dass der Weg von einem Molekül zu einem zugelassenen Medikament durch Drug Repurposing ein systematischer, datengetriebener Prozess ist, der evidenzbasierte Identifikation, präklinische Validierung, zielgerichtete klinische Evaluation und regulatorische Absicherung umfasst. Durch die kluge Nutzung vorhandener Substanzen können Patienten mit seltenen Erkrankungen schneller neue Behandlungsoptionen erhalten, ohne dass die gesamte Entwicklung von Grund auf neu beginnen muss.


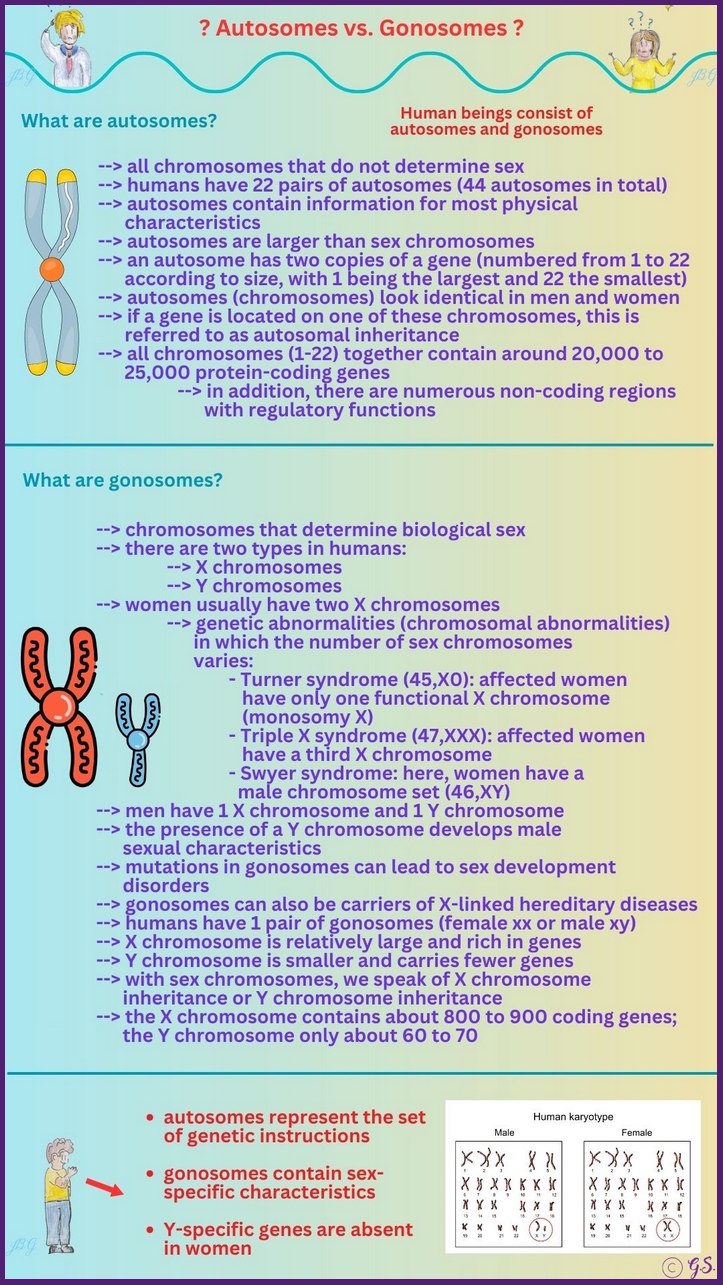
Autosomen vs. Gonosomen
Im Menschen liegt das genetische Material auf zwei Arten von Chromosomen: Autosomen und Gonosomen. Beide Chromosomenarten tragen wesentliche Informationen, die die Entwicklung, Funktion und Vielfalt des Organismus bestimmen.
Autosomen sind die ersten 22 Chromosomenpaare und tragen die alltäglichen Eigenschaften unseres Körpers, die nicht direkt mit der Geschlechtsausprägung verknüpft sind. Sie enthalten die Mehrzahl der Gene, die für Merkmale wie Hautfarbe, Blutgruppen, Stoffwechselwege und viele Organfunktionen verantwortlich sind.
Gonosomen, beim Menschen das 23. Paar, bestimmen das Geschlecht (bei Menschen typischerweise zwei X-Chromosomen bei Frauen und ein X- und ein Y-Chromosom bei Männern) und tragen zusätzlich Gene, die sowohl geschlechtsspezifische Merkmale als auch andere physiologische Eigenschaften beeinflussen können.
Der Mensch besitzt also sowohl Autosome als auch Gonosomen, deren Gene zusammen das Erbgut, die Entwicklung und die Funktion des Organismus prägen. Die Gesamtheit der Gene auf Autosomen und Gonosomen interagiert komplex, um die individuelle Entwicklung, die Heterogenität der Phänotypen und die Anfälligkeiten für verschiedene Erkrankungen zu formen.

EEG vs. qEEG
Das Elektroenzephalogramm (EEG) ist eine nicht-invasive Methode zur Messung der zeitlich hochauflösenden elektrischen Aktivität des Gehirns. Es liefert Rohdaten in Form von Spannungsänderungen, die über Elektroden auf der Kopfhaut aufgezeichnet werden und Einblicke in spontane oder reizinduzierte neuronale Prozesse geben.
In der klinischen Praxis dient das EEG überwiegend der Diagnose und Verlaufskontrolle von neurologischen und neuropsychiatrischen Erkrankungen, insbesondere Epilepsien, Schlafstörungen und Hirnfunktionen in verschiedenen Krankheitsstadien. In der Forschung hingegen wird das EEG nicht nur zur Beschreibung der normalen Gehirnaktivität genutzt, sondern auch als sensitive Marker für kognitive Prozesse, Schlafarchitekturen, Entwicklungs- und Alterungsprozesse sowie pathophysiologische Veränderungen unter verschiedenen Interventionen.
In der Forschung steht oft die Weiterentwicklung von methodischen Ansätzen wie standardisierte Preprocessing-Pipelines, Artifact-Entfernung, Querschnitts- und Längsschnitt-Analysen im Vordergrung und maschinelles Lernen eröffnet neue Möglichkeiten, EEG-Daten in robusten Biomarkern zu übersetzen.
Das qEEG (quantitative EEG) ergänzt das herkömmliche EEG, indem es aus den Rohsignalen systematische Merkmale extrahiert und diese in einer normativen Referenz vergleicht. Ziel von qEEG ist es, Muster und Abweichungen quantitative zu charakterisieren, die mit verschiedenen Zuständen oder Behandlungen assoziiert sein könnten. Dazu gehören Topographie-Analysen der EEG-Leistung über Schädeloberflächen hinweg, die Ableitung z-transformierter Werte, sowie Datenvisualisierungen wie Sortierung nach Norm- oder Abweichungsprofilen.
In der Forschung ermöglicht qEEG eine umfassendere Quantifizierung von neurophysiologischen Unterschieden zwischen Gruppen, Effekte von Interventionen oder Entwicklungsverläufe, wobei die Ergebnisse oft in Relation zu altersspezifischen oder methodisch bedingten Normen gesetzt werden.
In der Praxis bedeutet dies, dass Forschungsarbeiten sorgfältig definieren sollten, ob sie primär die qualitativ-zeitliche Dynamik des Hirns (EEG) oder die quantitative Abhandlung der Signale in normierter Form (qEEG) betonen, und wie die gewonnenen Befunde in Bezug zu bestehenden Modellen, Bildgebung und klinischen Befunden interpretiert werden.


Multi-Omics
Wichtigkeit von Omics in der Forschung
"Omics" bezeichnet einen Forschungszweig der Biowissenschaften, der sich mit der Untersuchung aller Moleküle eines bestimmten Typs in einem lebenden System befasst. Zu dieser Forschung gehören Techniken mit der Endung "-omics", wie beispielsweise die Genomik: die Untersuchung aller Gene eines Genoms.
Omics-Ansätze erfassen ganzheitliche Systeme auf molekularer Ebene statt einzelner Bausteine. Große Datensätze ermöglichen Mustererkennung, Biomarker-Profile, Krankheitspfade sowie individuelle Unterschiede (Personalisierung).
Sie liefern mechanistische Einsichten durch die Verknüpfung von Genen, Proteinen und Metaboliten, helfen beim Verständnis von Krankheitsursachen und Resistenzmechanismen und unterstützen die Biomarker-Entwicklung für Diagnose, Prognose und Therapieberatung; zudem können sie neue therapeutische Targets aufdecken, oft durch integrative Analysen verschiedener Omics-Ebenen.
In der Präzisionsmedizin ermöglichen sie die Anpassung von Therapien an individuelle molekulare Profile. Schließlich beschleunigen Hochdurchsatztechniken und bioinformatische Tools Hypothesentests und Replikationen über verschiedene Modelle hinweg.
Omics-Technologien sind in der Forschung essenziell, da sie die ganzheitliche Analyse biologischer Moleküle simultan ermöglichen, um komplexe Krankheiten auf molekularer Ebene zu verstehen. Omics-Technologien wird in der Medizin ein enormes Potenzial zugeschrieben. Multi-Omics Analysen gehören mittlerweile zu den grundlegenden Methoden in der präklinischen und klinischen Forschung, denn sie treiben die personalisierte Medizin voran.


Drug Repurposing vs. Off-Label
Drug Repurposing und Off-Label-Use unterscheiden sich klar in Ziel, Prozess und rechtlicher Einordnung.
Drug Repurposing (Wirkstoff-Umwidmung) bezeichnet den systematischen, wissenschaftlich orientierten Prozess, vorhandene Medikamente für neue Indikationen zu prüfen, zu entwickeln und idealerweise eine neue Zulassung anzustreben.
Ziel ist es, Nutzen, Sicherheit und Kosteneffizienz zu bewerten, um eine offizielle Genehmigung für die neue Indikation zu erhalten. Beispiele umfassen die Erforschung bekannter Substanzen für bislang ungeprüfte Krankheiten oder Patientengruppen, oft unterstützt durch beobachtete Wirkungen oder statistische bzw. computergestützte Ansätze. Im Verlauf des Repurposing wird die neue Indikation formal geprüft, Studien geplant und gegebenenfalls eine neue Zulassung erarbeitet.
Off-Label-Use beschreibt den Einsatz eines Arzneimittels außerhalb der behördlich genehmigten Indikation, Dosis oder Darreichungsform. Der Wirkstoff liegt in der passenden Form vor, wird jedoch zur Behandlung eines anderen Krankheitsbildes verwendet, für das keine offizielle Zulassung besteht.
Das Risiko besteht darin, dass Wirksamkeit und Sicherheit für die neue Indikation nicht umfassend geprüft sind. In der Praxis erfolgt Off-Label-Use oft auf ärztlicher Einzelentscheidung, mit variabler Kostenübernahme durch die Krankenkassen und je nach Rechtsordnung unterschiedlichen Haftungsfolgen. Beispiele sind der Einsatz eines Medikaments aus einer anderen Indikation oder eine abweichende Dosierung, die nicht in der Zulassungsdokumentation steht.
Off-Label-Use ist ein pragmatischer, rechtlich weniger geordneter Einsatz außerhalb der Zulassung, während Drug Repurposing ein geplanter, wissenschaftlich fundierter Prozess ist, der darauf abzielt, eine neue Indikation offiziell zuzulassen.


Apoptose
Apoptose, oder programmierter Zelltod, ist ein streng regulierter Prozess, bei dem sich Zellen auf kontrollierte und geordnete Weise bewusst selbst zerstören. Diese Selbstzerstörung dient als entscheidender Mechanismus, um beschädigte, funktionsunfähige oder unnötige Zellen zu beseitigen, ohne Entzündungen im umliegenden Gewebe hervorzurufen.
Sie spielt eine grundlegende Rolle bei der Entwicklung, indem sie Organismen ermöglicht, Organe zu formen und vorübergehende Zellpopulationen zu entfernen, sowie bei der Aufrechterhaltung der Gewebehomöostase, indem sie die Zellbildung und den Zelltod bei Erwachsenen ausgleicht.
In der Forschung ist die Apoptose von zentraler Bedeutung, da eine Fehlregulation dieses Prozesses mit einer Vielzahl von Krankheiten in Verbindung gebracht wird, darunter neurodegenerative Erkrankungen, Krebs, Autoimmunerkrankungen und Reaktionen auf Verletzungen.
Das Verständnis der Signalnetzwerke, die die Apoptose steuern – wie das Gleichgewicht zwischen Überlebens- und Todesignalen, der mitochondriale Weg und die Caspase-Kaskade – zeigt nicht nur, wie Zellen zwischen Leben und Tod entscheiden, sondern bietet auch Ansatzpunkte für therapeutische Interventionen, die darauf abzielen, das Überleben von Zellen bei degenerativen Erkrankungen zu fördern oder den Zelltod bei Krebs zu verstärken.


Genomische Variante
Genomische Varianten sind die subtilen Unterschiede in der DNA-Sequenz, die jeden Menschen genetisch einzigartig machen. Diese Variationen können Einfluss darauf haben, wie eine Person aussieht, wie ihr Körper funktioniert und wie sie auf Medikamente und Umweltfaktoren reagiert.
Die meisten Varianten sind häufig und harmlos und tragen zur normalen Vielfalt von Merkmalen wie Augenfarbe, Körpergröße und Stoffwechsel bei. Einige Varianten können jedoch biologische Prozesse stören und das Risiko für bestimmte Krankheiten erhöhen oder die Schwere der Symptome beeinflussen, wodurch sie für Diagnose, Prognose und Behandlungsentscheidungen klinisch relevant sind.
Einzelnukleotid-Polymorphismen (SNPs) sind die häufigste Art genetischer Variationen, bei denen es zu einer Veränderung eines einzelnen Nukleotids an einer bestimmten Position im Genom kommt. SNPs können helfen, Unterschiede in der Reaktion auf Medikamente, der Anfälligkeit für Krankheiten und den körperlichen Merkmalen zwischen Individuen zu erklären.
Während viele SNPs keine Auswirkungen haben, können andere die Genaktivität oder Proteinfunktion verändern und so zu Gesundheitsrisiken oder zum Schutz beitragen. Das Verständnis genomischer Varianten, einschließlich SNPs, hilft Forschern und Klinikern, zwischen normalen Variationen und Veränderungen zu unterscheiden, die möglicherweise ärztliche Hilfe erfordern, und ermöglicht so eine präzisere und personalisierte Versorgung.
In genetischen Berichten wird jede Variante anhand einer standardisierten, aber kontextabhängigen Notation beschrieben, wobei die genaue Formulierung je nach berichtendem Labor, Testpanel und klinischem Zweck variieren kann. Einige Berichte geben möglicherweise die genomische Position und Referenz-/Alternativallele an, andere listen möglicherweise die Veränderung auf der Ebene des Gens oder Transkripts auf, und wieder andere enthalten möglicherweise die damit verbundene klinische Bedeutung, die Häufigkeit in der Bevölkerung und Belege. Das bedeutet, dass dieselbe Variante in verschiedenen Berichten unterschiedlich beschrieben werden kann, was die Bedeutung der Konsultation eines Klinikers oder genetischen Beraters für eine einheitliche Interpretation der Ergebnisse unterstreicht.


Satelliten-DNA / Junk-DNA
Satelliten-DNA und Junk-DNA sind Begriffe, die häufig verwendet werden, um Teile des Genoms zu beschreiben, die einst als nicht funktionsfähig galten, jedoch wichtige Rollen spielen, die in der heutigen Forschung zunehmend anerkannt werden.
Satelliten-DNA bezieht sich auf hoch repetitive Sequenzen, die typischerweise in zentromeren und perizentromeren Regionen vorkommen, wo ihre Organisation zur Chromosomenstruktur, Stabilität und Segregation während der Zellteilung beiträgt.
Junk-DNA ist ein weiter gefasster Begriff, der historisch verwendet wurde, um Genomregionen ohne offensichtliches Protein-Kodierungspotenzial zu bezeichnen, aber heute umfasst er ein breites Spektrum an regulatorischen Elementen, nicht-kodierenden RNAs und anderen Sequenzmerkmalen, die die Genexpression, die Genomarchitektur und die Evolutionsdynamik beeinflussen.
Zusammen sind diese Genomregionen entscheidend für das Verständnis der Chromosomenbiologie, der Genomregulation und der Mechanismen der Evolution. Sie bieten auch potenzielle Einblicke in menschliche Krankheiten, da Variationen in repetitiven Elementen das Chromosomenverhalten und die Genregulation beeinflussen können. In der modernen Forschung stellt die erneute Betrachtung von Satelliten- und nichtkodierender DNA die veraltete Ansicht von nichtfunktionaler DNA in Frage und hebt deren Beitrag zur Zellfunktion, zu Entwicklungsprozessen und zur genomischen Plastizität hervor.


Basic, Preclinical und Translational Research
In der biomedizinischen Forschung bilden sich drei miteinander verzahnte Forschungsbereiche heraus, die gemeinsam den Weg von der grundlegenden Entdeckung bis zur klinischen Anwendung gestalten. Gemeinsam ermöglichen Basic, Preclinical und Translational Research eine systematische und iterative Entwicklung von wissenschaftlichen Erkenntnissen zu praktischen medizinischen Lösungen.
Die Grundlagenforschung, auch als Basic Research bekannt, widmet sich dem reinen Verständnis der zentralen Mechanismen biologischer Systeme. Sie zielt darauf ab, die Funktionsweisen von Genen, Proteinen und Zellprozessen zu entschlüsseln und neue Theorien bzw. Hypothesen zu formulieren, ohne dabei unmittelbar eine konkrete therapeutische Anwendung zu verfolgen. Die Erkenntnisse der Grundlagenforschung liefern das fundamentale Wissen, auf dem spätere Ansätze aufbauen können, und schaffen oft neue Fragestellungen, die den Forschungskorridor in Richtung Anwendung erweitern.
Die präklinische Forschung folgt dem Prinzip, aus theoretisch gewonnenem Wissen praktikable und sichere Interventionen abzuleiten. Sie testet Ideen zunächst in kontrollierten, nicht- menschlichen Systemen wie zellbasierten Modellen oder Tiermodellen, um Wirksamkeit, Sicherheit, Dosierung und toxikologische Profiles zu evaluieren. Ziel ist es, robuste Daten zu erzeugen, die eine sichere Weiterentwicklung in den klinischen Raum ermöglichen und den Übergang in erste menschliche Studien, typischerweise in Form von IND-Anträgen oder vergleichbaren Zulassungsverfahren, unterstützen. Die präklinische Phase dient somit als Brücke zwischen der Entdeckung im Labor und der Anwendung am Patienten.
Die Translational Research, oft als „Bench-to-Bedside“-Ansatz beschrieben, fokussiert sich darauf, Mechanismen und Erkenntnisse aus der Grundlagenforschung gezielt in klinische Anwendungen zu überführen. Sie integriert mehrere Disziplinen und Ebenen – von Proof-of-Concept-Studien über die Entwicklung translationaler Biomarker bis hin zu frühen klinischen Studien und kontinuierlichem Feedback aus der klinischen Praxis in die weitere Laborforschung. Ziel ist es, die Lücke zwischen Laborwissen und klinischer Relevanz zu schließen, Real-World-Faktoren zu berücksichtigen, regulatorische Anforderungen zu adressieren und letztlich neue Therapien schneller und sicherer in die Versorgung von Patientinnen und Patienten zu überführen.


Was sind Chaperone?
Chaperone sind wichtige molekulare Helfer in Zellen, die Proteine dabei unterstützen, sich in ihre richtige Form zu falten, sie stabilisieren, um Fehlfaltungen zu verhindern, und ihren Transport zu den richtigen Stellen in der Zelle steuern. Sie unterstützen neu gebildete Polypeptide dabei, sich zu ihrer korrekten dreidimensionalen Struktur zu falten und helfen bei dem Abbau fehlgefalteter Proteine. Sie arbeiten in einem hochgradig koordinierten Netzwerk mit anderen Qualitätskontrollsystemen zusammen, um sicherzustellen, dass Proteine ihre funktionellen Konformationen erreichen und aufrechterhalten, was für die Gesundheit und Homöostase der Zellen von entscheidender Bedeutung ist.
In der Forschung werden Chaperone untersucht, um Krankheiten zu verstehen, die auf Fehlfaltungen und Aggregationen von Proteinen beruhen, wie z. B. neurodegenerative Erkrankungen, und um grundlegende Prinzipien der Proteinbiologie, Faltungswege und der zellulären Stressreaktion aufzudecken. Durch die Aufklärung, wie Chaperone entfaltete oder teilweise gefaltete Proteine erkennen, mit ihnen über ATP-gesteuerte Zyklen interagieren und mit anderen zellulären Mechanismen zusammenarbeiten, können Wissenschaftler Strategien zur Modulation dieser Prozesse entwickeln, die potenzielle Anwendungen in der Therapeutik, Biotechnologie und im allgemeinen Verständnis der zellulären Qualitätskontrolle finden.
In der Forschung bieten sie Einblicke in fundamentale Prinzipien der Proteinfaltung, liefern Ansatzpunkte für Therapien gegen Faltungsstörungen und ermöglichen technologische Verbesserungen in Biotechnologie und therapeutischer Proteinproduktion. Chaperone können Off-Target-Effekte haben und nicht-funktionelle oder pathogene Konformationen in anderen Proteinen stabilisieren, was zu unbeabsichtigten zellulären Folgen führen kann. Es ist entscheidend, das richtige Maß an Stabilisierung zu erreichen.


Was sind SNAREopathien?
SNAREopathien sind Erkrankungen, die durch eine Funktionsstörung der SNARE-Proteine oder ihrer regulatorischen Partner entstehen und die Fusion synaptischer Vesikel sowie die Freisetzung von Neurotransmittern stören. SNAREs (Soluble NSF Attachment Protein Receptors) sind für die Vesikelfusion in Neuronen und anderen Zellen unerlässlich. Wenn ihre Funktion beeinträchtigt ist, kann dies zu Störungen der neuronalen Kommunikation und des zellulären Transports führen, was eine Reihe klinischer Symptome zur Folge hat.
Eine beeinträchtigte SNARE-Funktion kann die Freisetzung von Neurotransmittern verringern oder deregulieren und somit die synaptische Signalübertragung und die Funktion neuronaler Schaltkreise beeinträchtigen.
Ein ordnungsgemäßer Vesikeltransport ist für die Entwicklung, Aufrechterhaltung und aktivitätsabhängige Plastizität von Synapsen von entscheidender Bedeutung.
SNAREs kommen in vielen Geweben zum Einsatz, sodass SNAREopathien über das ZNS hinausgehende Multisystem-Symptome hervorrufen können.
Funktionsverlustmutationen (LOF) – wie bei einer STXBP1-Mutation – können die Effizienz der Vesikelfusion verringern und so die Freisetzung von Neurotransmittern senken.
Funktionsgewinnmutationen (GOF) oder dominant-negative Varianten (neuerdings auch bei STXBP1 entdeckt) können die Komplexbildung oder das Timing stören und zu einer abnormalen synaptischen Aktivität führen.
Mutationen können die Proteinstabilität, die Lokalisierung oder die Wechselwirkungen mit regulatorischen Partnern beeinträchtigen und zu zellulärem Stress oder veränderter Signalübertragung führen.
STXBP1 (Syntaxin-bindendes Protein 1) reguliert die Funktion des SNARE-Komplexes, der für die Fusion synaptischer Vesikel mit der Nervenzellmembran und die Freisetzung von Neurotransmittern von zentraler Bedeutung ist. Es bindet an Syntaxin-1 (STX1) und stabilisiert es in einer geschlossenen Konformation, wodurch eine vorzeitige Bildung des SNARE-Komplexes verhindert wird. Diese Kontrolle verhindert unangemessene Fusionsvorgänge. Während der Signalübertragung ermöglicht STXBP1 die ordnungsgemäße Bildung des SNARE-Komplexes mit VAMP2 (vesikuläres SNARE) und SNAP25 (Plasmamembran-SNARE), um den Vier-Helix-SNARE-Komplex zu bilden, der das Andocken, die Fusion und die Freisetzung von Neurotransmittern der Vesikel steuert. Varianten in STXBP1 können diese regulatorische Funktion stören, was zu einer beeinträchtigten oder dysregulierten SNARE-vermittelten Exozytose und daraus resultierenden neurologischen Entwicklungsstörungen und epileptischen Phänotypen führt.
Es gibt Forschungsergebnisse, die SNARE-Protein-Gene mit menschlichen Erkrankungen in Verbindung bringen, insbesondere mit seltenen neurologischen Entwicklungsstörungen und epileptischen Enzephalopathien.
Zu den wichtigsten Genen gehören STXBP1 (Syntaxin-bindendes Protein 1), STX1A, STX1B (Syntaxine), SNAP25, VAMP2 (Synaptobrevin-2), SCN-verwandte und andere interagierende Partner, die die Funktion des SNARE-Komplexes indirekt beeinflussen. Zu den Forschungsansätzen gehören Genetik/Genomik (Identifizierung pathogener Varianten), Funktionsstudien an Neuronen und Modellorganismen (z. B. Maus, Zebrafisch) sowie Untersuchungen zu potenziellen Therapien, die die synaptische Übertragung modulieren oder die Funktion des SNARE-Komplexes stabilisieren. Genetische Tests – insbesondere gezielte Panels oder Exomsequenzierung – können pathogene Varianten in SNARE-Genen wie STXBP1, STX1A, STX1B, SNAP25 und VAMP2 identifizieren.


Nonverbal sein mit STXBP1
Nonverbal bedeutet typischerweise, dass eine Person keine gesprochene Sprache zur Kommunikation verwendet. Sie kann auf alternative Kommunikationsweisen zurückgreifen, wie Gesten, Blickkontakt, Mimik, Bilder, Gebärdensprache oder unterstützende Technologien. Nonverbal ist ein Spektrum: Einige Personen sprechen nur sehr wenig oder inkonsistent, während andere möglicherweise überhaupt keine funktionale Sprache entwickeln.
Nonverbale Ergebnisse bei STXBP1-Varianten sind mit neuroentwicklungsbezogenen Störungen verbunden, darunter epileptische Enzephalopathien, intellektuelle Beeinträchtigungen und Sprach-/Sprachentwicklungsstörungen. Der nonverbale Status kann entstehen durch: globale Entwicklungsverzögerung, die Sprache und Motorik betrifft; Belastung durch Krampfanfälle und epileptische Aktivität, die Sprachnetzwerke beeinträchtigt; motorische Sprechstörungen (Apraxie oder Dysarthrie), die gesprochene Äußerung einschränken; kognitive Beeinträchtigungen, die expressive Sprachfähigkeit reduzieren; gleichzeitiges Auftreten von Autismus-Spektrum-Störungen, die Kommunikation beeinflussen.
Therapien und Interventionen können helfen und werden individuell auf die Fähigkeiten abgestimmt, mit Fokus auf alternative und augmentative Kommunikation (AAC) falls erforderlich. Augmentative and Alternative Communication (AAC) sind Geräte oder Methoden zur Unterstützung der Kommunikation, wie einfache Bildertafeln, Kommunikations-Apps, Eye-Gaze-Systeme, sprachgenerierende Hilfen in leichter oder anspruchsvoller Ausführung. Frühe intensive Interventionen sind besonders wichtig in Entwicklungsfenstern für Sprach- und Kommunikationsfähigkeiten. Die Kontrolle von Krampfanfällen kann in einigen Fällen die kognitiven und kommunikativen Ergebnisse verbessern.
Es gibt keine Heilung für STXBP1-assoziierte Erkrankungen, jedoch wird laufend zu zielgerichteten Therapien geforscht, die neuronale Schaltkreise beeinflussen. Es gibt umfangreiche Forschung zu nonverbalen Kommunikationsformen bei neuroentwicklungsbedingten Störungen, auch im Kontext von STXBP1-assoziierten Erkrankungen. Zentrale Forschungsfelder sind die Zusammenhänge zwischen Hirnnetzwerken, Sprache und Kommunikation zu untersuchen, und wie Krampfkontrolle und neuronale Aktivitätsmuster kognitive und sprachliche Entwicklung beeinflussen können. Spezifisch zu STXBP1 fokussieren sich Untersuchungen auch darauf, Frühintervention und AAC zu optimieren, um die Kommunikationsmöglichkeiten zu maximieren.


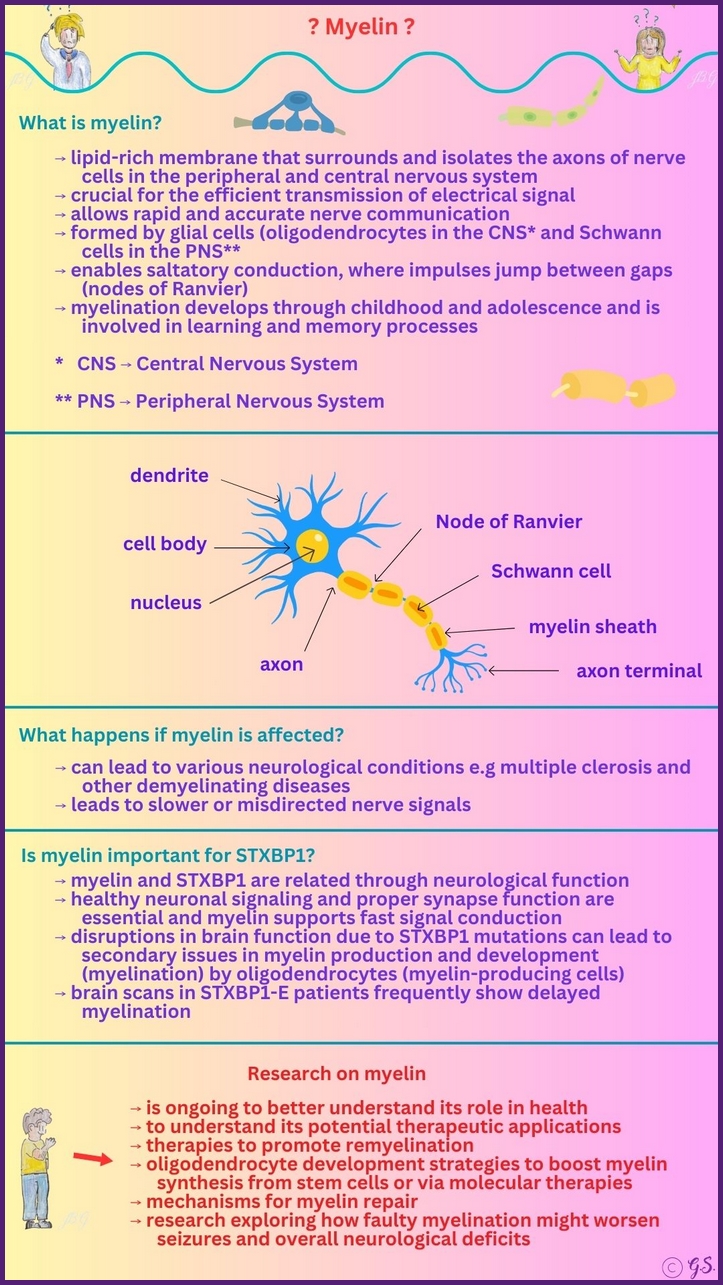
Schützende Hülle: Myelin
Myelin ist eine schützende Fettschicht, die Axone im Nervensystem schützt und umhüllt. Sie wird von Gliazellen gebildet (Oligodendrozyten im ZNS und Schwann-Zellen im PNS) und erhöht die Geschwindigkeit sowie Stabilität der Nervenleitung. Myelin wirkt wie eine Isolationsschicht, die die Übertragung der Nervenimpulse beschleunigt und die Signalstörung reduziert. Myelin ermöglicht eine saltatorische Fortleitung, bei der Impulse zwischen den Schnürringen springen, wodurch die Übertragung deutlich schneller wird.
Demyelinisierung bedeutet Verlust oder Schädigung der Myelinschicht, was zu langsameren oder falsch geleiteten Signalen führt. Klinisch kann dies zu Schwäche, Koordinationsproblemen, Taubheit, Sehstörungen und Muskelkrämpfen führen; abhängig davon, welche Nerven betroffen sind, können Erkrankungen wie Multipler Sklerose (MS) oder andere demyelinisierende Krankheiten auftreten.
STXBP1 (Syntaxin-binding Protein 1) kodiert ein Protein, das eine wichtige Rolle bei der synaptischen Übertragung und der Freisetzung von Neurotransmittern spielt. Es ist primär kein Ziel der Myelinisierung. Dennoch ist eine gesunde neuronale Signalübertragung und eine ordnungsgemäße Synapsenfunktion wesentlich für die Entwicklung und Funktion des ZNS, und Myelinisierung unterstützt eine schnelle Leitfähigkeit, die normales neuronales Aktivitätsmuster ermöglicht.
Forschung an Myelin ist extrem wichtig für STXBP1-Enzephalopathie, da das STXBP1-Gen eine Schlüsselrolle bei der Funktion von Nervenzellen spielt, einschließlich der Signalübertragung und möglicherweise der Myelinbildung oder -funktion, was zu Entwicklungsverzögerungen, Epilepsie und Bewegungsstörungen führt. Da STXBP1 die Synapsenreifung beeinflusst, ist das Verständnis der Myelinisierung essenziell, um die neurologischen Defizite zu verstehen und neue Therapien zu entwickeln, die die Kommunikation zwischen Nervenzellen verbessern könnten.
Aktuelle Ansätze fokussieren sich auf die Differenzierung von oligodendrozytären Vorläuferzellen aus Stammzellen, die Förderung der Reifung zu Myelin-produzierenden Oligodendrozyten sowie die Optimierung der Remyelinisierung. Die Identifikation genetischer Variationen, die die Myelinisierung beeinflussen, bietet Ansatzpunkte für personalisierte Therapien und Strategien zur Remyelinisierung. Aktuelle Forschung untersucht, wie STXBP1-genetische Varianten oder Funktionsstörungen die neuronale Aktivität beeinflussen und ob diese Veränderungen die Myelinbildung oder Remyelinisierung indirekt beeinflussen künnten.

Modifikatorgene
Gene spielen eine große Rolle dabei, wer wir sind. Der Genotyp ist die Gesamtheit der Gene, die wir von unseren Eltern erben, während der Phänotyp unser tatsächliches Aussehen oder die Funktionsweise unseres Körpers beschreibt.
Manchmal können andere Gene, sogenannte Modifikatorgene, diese Merkmale beeinflussen, indem sie sie verstärken, abschwächen oder ihr Erscheinungsbild verändern. Selbst wenn zwei Menschen ähnliche Hauptgene haben, können Modifikatorgene zu Unterschieden in ihren Merkmalen führen. Dies zeigt, wie komplex die Genetik sein kann und warum Menschen unterschiedlich aussehen oder sich unterschiedlich verhalten können, obwohl sie viele der gleichen Gene haben.
Modifikatorgene tragen so zur Komplexität genetischer Merkmale bei. Bei seltenen Krankheiten bestimmt der Genotyp einer Person – also ihre genetische Veranlagung – nicht immer direkt ihren Phänotyp, also die beobachtbaren Merkmale oder Symptome. Personen mit derselben Mutation können unterschiedliche Symptome aufweisen, und ähnliche Symptome können unterschiedliche genetische Ursachen haben. Umweltfaktoren und andere genetische Einflüsse beeinflussen häufig die Expression von Genen, wodurch die Beziehung zwischen Genotyp und Phänotyp komplex und nicht immer eindeutig ist.
Das Erkennen dieser fehlenden Eins-zu-Eins-Beziehung ist entscheidend für eine genaue Diagnose und personalisierte Behandlungsansätze bei seltenen Krankheiten, wie STXBP1-RD. Modifier-Gene können einen entscheidenden Einfluss darauf haben, wie sich eine STXBP1-Mutation im Laufe des Lebens manifestiert, was die Komplexität der Erkrankung erklärt und die personalisierte Therapie vorantreibt. Sie erklären, warum STXBP1-Betroffene so unterschiedliche Symptome und Schweregrade zeigen, obwohl sie die gleiche Gen-Mutation haben.


Stammzellen vs. Progenitorzellen
Stammzellen, die ultimativen "Meisterzellen" (Totipotente, pluripotente oder adulte Stammzellen), sind Zellen mit der Fähigkeit zur selbständigen Vermehrung und zur Differenzierung in verschiedene Zelltypen des Organismus. Sie können sich unbegrenzt erneuern und, je nach Typ, zu vielen oder sogar allen Zellarten des Körpers entwickeln.
Progenitoren/Prekursoren sind ihre Nachkommen, die bereits einer bestimmten Linie zugeordnet sind, über eine begrenzte Selbsterneuerung und ein eingeschränktes Differenzierungspotenzial verfügen (oligopotent oder unipotent).
Progenitorzellen sind also Vorläuferzellen, die sich bereits in eine begrenzte Anzahl verschiedener Zelltypen differenzieren konnten. Ihre Teilungsfähigkeit ist oft stärker eingeschränkt als die von Stammzellen, und sie verlieren mit der Zeit ihre Fähigkeit zur unbegrenzten Selbst-Erneuerung.
Prekursorzellen ist ein weiter gefasster Begriff, der Zellen beschreibt, die sich auf dem Weg der Differenzierung befinden und dem Endtypus bereits näherstehen, aber sich noch nicht in die endgültige reife Zelle ausgebildet haben. Prekursorzellen können sowohl Progenitorzellen als auch differentiell stärker eingeschränkte Zustände umfassen.
Kurz gesagt: Stammzellen können sich unbegrenzt erneuern und in viele Zelltypen differenzieren, Progenitorzellen können sich nur in eine begrenzte Reihe verwandter Zelltypen differenzieren und sich begrenzt vermehren, und Prekursorzellen bezeichnen allgemein Zellen kurz vor der endgültigen Spezifizierung auf einen bestimmten Zelltyp.


Was ist ein Spleißosome?
Ein Spleißosom ist ein großes, enzymatisches Molekülkomplex aus Proteinen und RNA im Zellkern von Eukaryoten, der das Spleißen katalysiert. Seine zentrale Aufgabe dieser molekulare Maschine ist das Entfernen von störenden Introns (nicht-kodierende Abschnitte) aus dem prä-mRNA-Strang und das anschließende Zusammenfügen der verbleibenden Exons (kodierenden Abschnitte) zu einer reifen Messenger-RNA, die dann als Vorlage fü die Proteinsynthese dient. Es besteht aus kleinen nukleären Ribonukleinsäuren (snRNAs) und über 100 Proteinen, die sich dynamisch zu einer komplexen Struktur zusammenfügen.
Dieser Prozess des Spleißens ist essenziell, weil er sicherstellt, dass nur korrekt zusammengesetzte mRNA-Mekteile den Zellkern verlassen und in Proteine übersetzt werden. Die Funktionsweise beruht auf der Erkennung spezieller Spleißsignale an den Grenzen von Introns und Exons sowie auf der katalytischen Aktivität der RNA-Komponenten des Spleißosoms, die die Spaltung und Verknüpfung der RNA durchführen. Fehler im Spleißen führen zu fehlerhaften Proteinen oder zu verlorener Proteinexpression und sind mit vielen Erkrankungen assoziiert, unter anderem genetischen Speicherkrankheiten, einigen Krebsarten und neurodegenerativen Störungen.
Ohne das Spleißosom könnte eine reife mRNA nicht entstehen. In der Forschung ist das Spleißen deshalb ein wichtiger Regulierungspunkt der Genexpression und der korrekten Synthese von Proteinen, weshalb modulare oder krankheitsbezogene Spleißveränderungen gezielt untersucht und potenziell therapeutisch adressiert werden.


Was ist ein Register?
Register sind für die STXBP1-bezogene Forschung von entscheidender Bedeutung, da sie systematisch standardisierte Längsschnittdaten aus verschiedenen Standorten sammeln. Dies ermöglicht ein klareres Bild vom natürlichen Verlauf der Erkrankung, einschließlich Ausbruch, Fortschreiten und Variabilität der Symptome. Durch die Erfassung von Genotyp-Phänotyp-Beziehungen, Funktionsdaten und Ergebnissen im Zeitverlauf unterstützen Register die evidenzbasierte klinische Entscheidungsfindung und personalisierte Behandlungspläne. Sie erleichtern auch aussagekräftige Vergleiche zwischen verschiedenen Zentren, was besonders für seltene Erkrankungen wichtig ist, bei denen einzelne Zentren nur eine begrenzte Anzahl von Fällen sehen.
Zu den wichtigsten Vorteilen gehören harmonisierte Datenelemente und Kodierungen, die eine Zusammenführung und Vergleichbarkeit zwischen Studien ermöglichen. Prospektive und retrospektive Dateneingaben aus verschiedenen Quellen (Kliniken, Labore, von Patienten gemeldete Informationen) maximieren die Vollständigkeit. Register verbessern die Aussagekraft und Durchführbarkeit von Beobachtungsstudien und erhöhen das Potenzial für die Teilnahme an interventionellen Studien. Sie verbessern das Verständnis des natürlichen Krankheitsverlaufs, liefern Informationen zu geeigneten Endpunkten und beleuchten Variabilitäten im Verlauf und in der Prognose. Die Datenqualität wird durch standardisierte Validierung, sorgfältigen Umgang mit fehlenden Daten und eine robuste Governance gestärkt.
Letztendlich übertragen Register Forschungsfortschritte in die klinische Praxis und Politik, indem sie zuverlässige, standortübergreifende Evidenz liefern, die die Entwicklung von Leitlinien, die Ressourcenplanung und die patientenzentrierte Versorgung unterstützt.


Vektor vs. Kapsid
Ein Vektor in der Biomedizin ist ein Trägersystem, das Gene oder genetische Informationen in Zielzellen transportiert. Vektoren können auf verschiedenen Grundlagen beruhen, zum Beispiel virale Vektoren, die Kapside als Träger nutzen, oder nicht-virale Vektoren wie Plasmide, liposomale Systeme oder Nanopartikel. Der Zweck eines Vektors besteht darin, eine bestimmte genetische Ladung sicher und zielgerichtet in Zellen zu liefern, oft mit therapeutischem oder forschungsbezogenem Ziel.
Ein Kapsid (Capsid) ist die proteine Hülle eines Virus, die das genetische Material schützt und die Form des Partikels bestimmt. Es besteht aus wiederholten Proteinuntereinheiten (Kapsomeren) und dient primär dem Schutz, der Stabilität und der Verpackung des viralen Genoms.
Der wesentliche Unterschied ist, dass das Kapsid eine Struktur des Virus ist, die das virale Genom schützt und transportiert, während der Vektor ein Trägersystem ist, das genutzt wird, um genetische Informationen in Zellen zu übertragen. Ein viraler Vektor verwendet oft ein Kapsid als Träger, aber nicht alle Vektoren basieren auf viralen Kapsiden; nicht-virale Vektoren verwenden andere Trägerformen.
In der Forschung ist der Vektor der gesamte Transportmechanismus (oft ein modifiziertes Virus), der genetisches Material in eine Zelle bringt, während das Kapsid die schützende Proteinhülle bildet, die das genetische Material umgibt und der Zelle hilft, zu erkennen, in welche Zelle sie eindringen soll. Das Kapsid ist demnach ein Bestandteil des Vektors.
Der Vektor wird gentechnisch so modifiziert, dass er das gewünschte Gen transportiert und sich nicht repliziert, während das Kapsid bzw. seine Hülle angepasst wird, um spezifische Zellen gezielt anzusteuern. In der Entwicklung von Vektoren werden Kapside oft verändert oder ausgetauscht, damit sie nur die gewünschten Zellen adressieren. Ein Vektor besteht demnach aus dem genetischen Material (dem zu transportierenden Gen) und dem Träger, oft ein modifiziertes Virus (wie Adenovirus oder Lentivirus) oder ein Plasmid. Virale Vektoren werden so modifiziert, dass sie sich nicht replizieren können und das Zielgen freisetzen.
Ein Plasmid ist ein kleines, ringförmiges DNA-Molek&uum;, das unabhängig vom Chromosom einer Zelle existieren kann. Plasmide tragen oft nur wenige Gene. In der Biotechnologie werden Plasmide als Vektoren genutzt, um genetische Informationen in Zellen zu transportieren, Gene zu klonieren oder Proteine zu produzieren.


DNA, RNA und Chromosome
Chromosomen, DNA & RNA: Die Bausteine unseres Erbguts.
Molekulare Grundlagen des Lebens beginnen bei den Chromosomen, der DNA und der RNA – den Bausteinen unseres Erbguts. Chromosomen sind die Träger der genetischen Information und dienen als Lagerorte für lange DNA-Fäden, die die Baupläne aller Lebewesen enthalten. Diese DNA wiederum umfasst die Gene, die die Anweisungen liefern, wie Proteine aufgebaut werden sollen, die eine Vielzahl von Funktionen im Organismus erfüllen. Die chemische Information in der DNA wird in eine passende Vorlage überführt, indem sie in RNA transkribiert wird. Diese RNA dient als Botenmolekül, das die genetische Information aus dem Zellkern zu den Ribosomen transportiert, wo die eigentliche Proteinsynthese stattfindet – die Translation. Dabei werden aus den Bausteinen der Aminosäuren Proteine zusammengesetzt, deren Dreidimensionalität und Struktur maßgeblich ihre Funktionen bestimmen.
Die DNA besitzt bemerkenswerte Eigenschaften: Sie speichert Informationen zuverlässig, ermöglicht Kopien für die Zellteilung und kann durch Mutationen neue Eigenschaften hervorrufen, die sich auf Entwicklung, Gesundheit und Vererbung auswirken können. RNA hingegen bietet Flexibilität und Regulierung, fungiert als Vermittlerin bei der Ablesung der Bauanleitung und steuert zeitliche Abläufe sowie die Feinabstimmung der Genexpression. Zusammen erklären Chromosomen, DNA und RNA, wie Erbinformationen vererbt werden, wie Zellen funktionieren, wie Organismen wachsen, wie Arten sich unterscheiden und wie Veränderungen im genetischen Code zu Phänotypen führen können.


Meine, deine DNA
DNA, oder Desoxyribonukleinsäure, ist der grundlegende Bauplan des Lebens und enthält die genetischen Anweisungen, die die Eigenschaften aller lebenden Organismen bestimmen. Sie besteht aus zwei Strängen, die eine Doppelhelixstruktur bilden und Sequenzen von Nukleotiden enthalten, die genetische Informationen kodieren. Dieser genetische Code ist in Genen organisiert, die die Synthese von Proteinen steuern, die für Lebensprozesse unerlässlich sind.
Zusätzlich zur Kern-DNA, die sich im Zellkern befindet, enthalten unsere Zellen auch mitochondriale DNA (mtDNA), die in den Mitochondrien – den energieproduzierenden Strukturen innerhalb der Zellen – gespeichert ist. Mitochondriale DNA ist ein kleines, ringförmiges Molekül, das sich in den Mitochondrien befindet – spezialisierten Organellen, die als Kraftwerke der Zelle bekannt sind. Im Gegensatz zur Kern-DNA wird mtDNA ausschließlich von der Mutter vererbt und enthält Gene, die in erster Linie an der Energieproduktion durch oxidative Phosphorylierung beteiligt sind.
Beide Arten von DNA spielen eine entscheidende Rolle in der Genetik, Vererbung und bei biologischen Funktionen und sind für die Forschung von entscheidender Bedeutung, da sie unterschiedliche, aber sich ergänzende Erkenntnisse liefern. Die Kern-DNA hilft uns, Merkmale, Krankheiten und Vererbung zu verstehen. Die mütterlich vererbte mitochondriale DNA ist nützlich, um die mütterliche Abstammungslinie zurückzuverfolgen, evolutionäre Beziehungen zu untersuchen und energiebezogene Krankheiten zu verstehen. Zusammen ermöglichen sie ein umfassenderes Verständnis biologischer Prozesse, Krankheitsmechanismen und Abstammung.

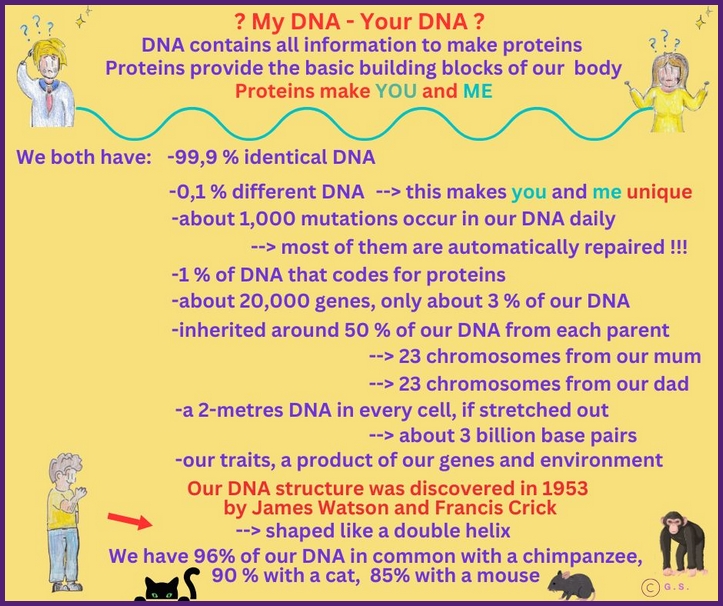
Was sind Proteine?
Proteine sind zentrale Bausteine des Lebens, essentielle Moleküle in allen lebenden Organismen, die für den Aufbau von Strukturen, Enzymen und die Regulierung biologischer Prozesse verantwortlich sind. Sie erfüllen in Zellen eine Vielzahl von Funktionen: Sie beschleunigen biochemische Reaktionen als Enzyme, unterstützen Struktur und Form von Geweben, transportieren Moleküle durchs Blut und kontrollieren den Ablauf biologischer Prozesse. Proteine bestehen aus langen Ketten von Aminosäuren, die sich in einer bestimmten Reihenfolge zu komplexen dreidimensionalen Strukturen falten. Diese Struktur bestimmt ihre Funktion: Kleine Veränderungen in der Abfolge oder falsches Falten können die Wirkung eines Proteins stark beeinträchtigen.
Proteine entstehen durch den Prozess der Proteinsynthese. Die Proteinsynthese ist der Prozess, bei dem Zellen auf der Grundlage der genetischen Informationen in der DNA Proteine produzieren. Dieser Prozess umfasst die Transkription der DNA zu RNA und der anschließenden Translation der RNA in eine Aminosäurekette. Die richtige Zusammensetzung und das richtige Zusammenspiel von Proteinen sind entscheidend für Gesundheit und Krankheit, Entwicklung und Verhalten. Das Verständnis von Proteinen hilft zu erklären, wie Zellen arbeiten, wie Organismen wachsen und wie Therapien gezielter entwickelt werden können. Das Verständnis ist für die Forschung von entscheidender Bedeutung, da es hilft, die Funktionsweise des Lebens zu verstehen, medizinische Behandlungen zu entwickeln und genetische Krankheiten zu erforschen.




Was sind SNPs?
Winzige Variationen in unserer DNA können alles beeinflussen, von unserer Gesundheit bis zu unserem Aussehen.
Einzelnukleotid-Polymorphismen, oder SNPs (ausgesprochen "Snips"), sind die häufigste Art genetischer Variationen unter Menschen. Es handelt sich dabei um Veränderungen einzelner Buchstaben in der DNA-Sequenz, die sich darauf auswirken können, wie Gene funktionieren und wie wir auf Krankheiten, Umweltfaktoren und Medikamente reagieren.
SNPs machen jeden von uns einzigartig. Diese Variationen können jedoch die Absorption, Verteilung, den Metabolismus und die Ausscheidung (ADME) von Medikamenten sowie deren Wirkziele beeinflussen.
Durch die Analyse dieser SNPs mittels pharmakogenetischer Tests können Gesundheitsdienstleister die wirksamsten und sichersten Medikamentendosen für jeden Einzelnen bestimmen.
Das Verständnis von SNPs hilft Wissenschaftlern, die Geheimnisse der menschlichen Vielfalt zu entschlüsseln und personalisierte Medizin und Gesundheitsversorgung zu entwickeln.


Was sind Gene?
Gene sind die grundlegendsten Bausteine der Vererbung und tragen die Informationen, die das Wachstum, die Entwicklung und die Funktionsweise eines Organismus steuern. Sie befinden sich auf Chromosomen und bestehen aus DNA. Die DNA enthält Sequenzen von Nukleotiden, die als Baupläne für Proteine und RNA-Moleküle dienen. Proteine übernehmen eine Vielzahl von Funktionen im Körper, darunter Strukturgebung, Enzymaktivität, Transport, Signalübermittlung und Immunabwehr. RNA-Moleküle spielen ebenfalls zentrale Rollen bei der Umsetzung genetischer Informationen in Funktionsprodukte, insbesondere bei der Regulation von Genaktivität und der Proteinsynthese.
Gene werden durch den Prozess der Genexpression genutzt. Zunächst wird die DNA in eine Abfolge von RNA-Schnipseln transkribiert, ein Prozess, der als Transkription bezeichnet wird. Die mRNA (Boten-RNA) dient dann als Vorlage für die Synthese von Proteinen in den Ribosomen, ein Vorgang, der als Translation bekannt ist. Dieser Ablauf wird durch eine Vielzahl von Regulatoren gesteuert, darunter Transkriptionsfaktoren, Epigenetik, RNA-Interferenz und nicht kodierende RNAs, die die Genaktivität erhöhen oder verringern können.
Die Genetik umfasst unterschiedliche Ebenen, Monogene Erkrankungen, Polygene Veranlagung und Genomische Variation. Monogene Erkrankungen entstehen durch Mutationen in einem einzelnen Gen und folgen oft klaren Erbgangslinien wie autosomal dominant, rezessiv oder X-chromosomal. Polygene Veranlagung bedeutet, dass viele Gene mit kleinen Einzelwirkungen zusammenkommen und in Kombination mit Umweltfaktoren das Risiko für komplexe Erkrankungen erhöhen. Genomische Variation umfasst Unterschiede in der DNA zwischen Menschen, wie einzelne Nukleotidpolymorphismen (SNPs), Insertionen, Deletionen oder Copy-Number-Variationen (CNVs). Diese Variationen können das Risiko für Erkrankungen beeinflussen, Merkmalsausprägungen modulieren oder die Reaktion auf Umweltfaktoren und Medikamente verändern.
Mutationen können zufällig auftreten oder vererbt werden. Sie können zu Verlust- oder Gewinnfunktionen von Genprodukten führen. Manche Mutationen sind neutral, andere schädlich oder vorteilhaft und können unter bestimmten Umweltbedingungen selektiv auftreten. In der medizinischen Praxis werden genetische Tests genutzt, um Mutationen, Prädiktionsmarker oder Trägerstatus zu identifizieren. Diese Tests reichen von Molekulargenetik über karyotypische Analysen bis hin zu modernen Sequenziermethoden wie Whole-Genome- oder Whole-Exome-Sequencing, die es ermöglichen, eine breite Palette genetischer Variationen zu entdecken.

Was ist ein Genom?
Ein Genom des Menschen ist die Gesamtheit des genetischen Materials in einer menschlichen Zelle und umfasst die komplette Sequenz der DNA sowie alle Gene und nicht kodierenden Bereiche, die Informationen für Entwicklung, Funktion, Reproduktion und Anpassung liefern. Beim Menschen besteht das Genom aus 22 autosomalen Chromosomenpaaren, dem X- und dem Y-Chromosom (bei Männern), sowie einer Vielzahl von regulatorischen Elementen, repetitiven Sequenzen und nicht kodierenden Regionen. Insgesamt enthält das Genom rund 3,2 Milliarden Nukleotidbasen und ungefähr 20.000 bis 25.000 Protein-codierende Gene, zusätzlich eine Vielzahl von RNA-Gengen, die nicht in Proteine übersetzt werden, sondern Funktionen wie Regulation, Splicing oder Ribosomenaufbau übernehmen.
Auf molekularer Ebene ist das menschliche Genom in Coding-Regionen (Exons), die Proteine oder funktionelle RNA liefern, und Nicht-Coding-Regionen unterteilt. Letztere umfassen Promotoren, Enhancer-Elemente, Silencer, Intronabschnitte, repetitives DNA-Material und zahlreiche regulatorische Sequenzen, die Sprache der Genexpression bestimmen. Die Genomorganisation wird durch Chromosomenstrukturen bestimmt und durch epigenetische Markierungen wie DNA-M methylierung oder Histonmodifikationen beeinflusst, welche die Aktivität einzelner Gene verändern, ohne die Sequenz selbst zu verändern.
Die Analyse des menschlichen Genoms umfasst das Sequenzieren, Annotieren und Interpretieren der Daten. Durch Genome-Wide-Association-Studien (GWAS) lassen sich Zusammenhänge zwischen genetischen Varianten, wie SNPs, und Merkmalen oder Krankheiten finden. Weitere Methoden wie Whole-Genome-Sequencing (WGS) oder Whole-Exome-Sequencing (WES) ermöglichen es, individuelle genetische Unterschiede zu identifizieren, die mit Erkrankungsrisiken, Pharmakogenetik oder Eigenschaftsausprägungen verbunden sein können.
Das menschliche Genom dient als Grundlage für personalisierte Medizin, in der Therapien, Medikamentenwahl und Präventionsstrategien auf das genetische Profil eines Individuums abgestimmt werden. Es erklärt auch unsere Verständnisse von Evolution, Verwandtschaftsbeziehungen und biologischer Vielfalt. Gleichzeitig wirft die Nutzung genomischer Daten ethische, rechtliche und soziale Fragen auf, etwa zu Datenschutz, genetischer Diskriminierung oder informierter Zustimmung.

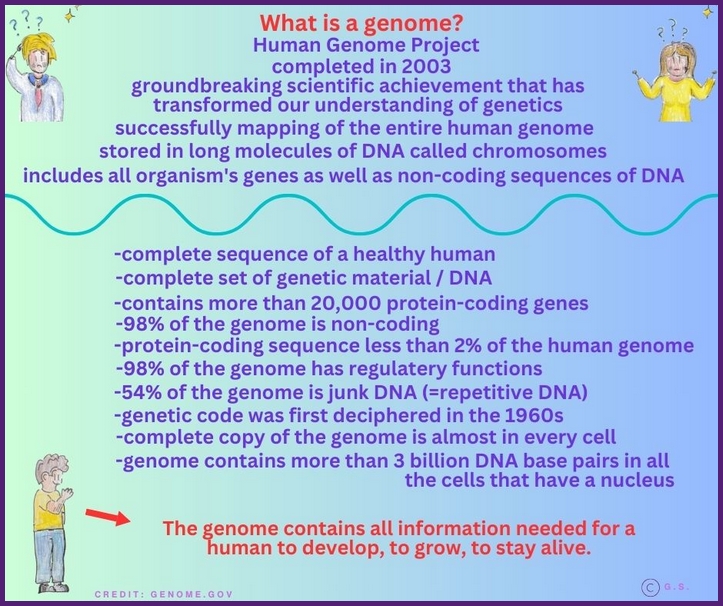
ASS vs. ADHS
Autismus-Spektrum-Störung (ASS) und ADHS unterscheiden sich grundlegend in der Ausrichtung ihrer Kernprobleme.
ASS ist eine Neuroentwicklungsstörung mit Fokus auf Kommunikation, soziale Interaktion und wiederholende Verhaltensweisen. Oft sind sensorische Empfindlichkeiten und eindimensionale oder stark fokussierte Interessen kennzeichnend.
ADHS dagegen ist durch Unaufmerksamkeit, Hyperaktivität und Impulsivität gekennzeichnet, wobei das Sozialverhalten zwar betroffen sein kann, aber nicht primär wie bei ASS.
Die Symptome bei ASS umfassen Verzögerungen in der Sprachentwicklung, Schwierigkeiten beim Blickkontakt, ein geringes Interesse an anderen Menschen sowie restriktive und repetitiven Muster. Beim ADHS dominieren Unaufmerksamkeit, leichtes Abgelenktsein, Vergesslichkeit, sowie motorische Unruhe und Impulsivität.
In sozialen Interaktionen kann es bei ADHS zu Schwierigkeiten beim Zuhören oder häufigerem Unterbrechen kommen, während bei ASS das soziale Verhalten stärker durch Verständnis- und Interaktionsschwierigkeiten geprägt ist.
Behandlungstechnisch konzentriert sich ASS vor allem auf Frühförderung, Verhaltenstherapie, Sprach- und Kommunikationstherapie sowie Ergotherapie. Unterstützung in Schule und Bildung, eine sensorikangepasste Umwelt und gegebenenfalls Behandlung begleitender Probleme wie Schlafstörungen oder Angst können ebenfalls Teil der Therapie sein; eine medikamentöse Behandlung zielt in der Regel nicht darauf ab, ASS selbst zu heilen, sondern begleitende Probleme zu behandeln. Bei ADHS kommen verhaltenstherapeutische Ansätze, Training in Schulleistungen sowie Organisations- und Planungstraining zum Einsatz. Medikamente, insbesondere Stimulanzien, sind in vielen Fällen hilfreich, wobei die genaue Wahl individuell angepasst wird. Unterstützung in Schule, Familienberatung und Psychoedukation gehören ebenfalls zum Behandlungskonzept.
In der Genetik zeigen sich bei beiden Störungen erhebliche Anteile hereditärer Faktoren. ASS hat einen komplexen, polygenen Beitrag mit vielen Risiko-Genen, deren Effekte klein sind; Umweltfaktoren spielen ebenfalls eine Rolle. Monogene Ursachen sind selten, aber familiäre Häufigkeit ist erhöht. Auch ADHS hat starke genetische Anteile, wobei ungefähr die Hälfte der beobachtbaren Varianz genetisch bedingt sein kann; hier dominieren polygenetische Einflüsse, wiederum kombiniert mit Umweltfaktoren. Beide Störungen zeigen Überschneidungen in bestimmten Merkmalen, wie Aufmerksamkeitsprobleme, soziale Herausforderungen und den Bedarf an individuell angepassten Unterstützungsmaßnahmen.

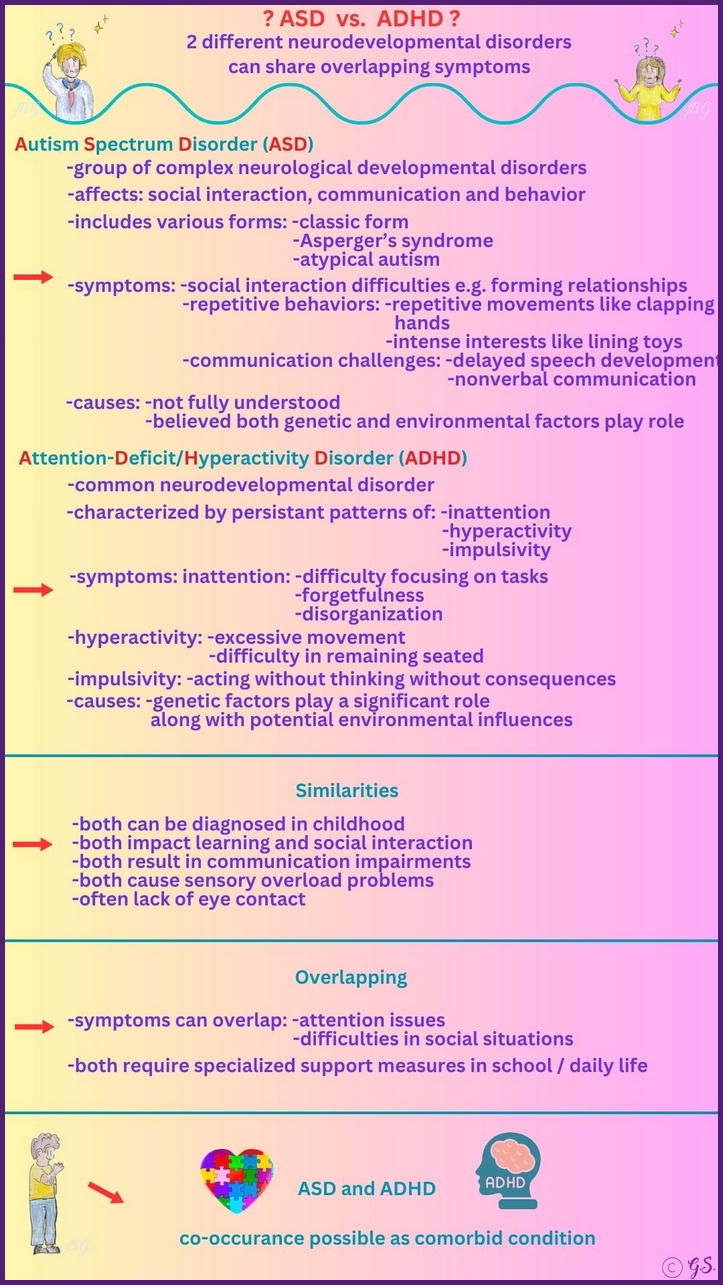
Was ist Autismus?
Autismus-Spektrum-Störung (ASS) ist eine neurologische Entwicklungsstörung, die sich bereits im frühen Kindesalter zeigt. Häufige Anzeichen können Unterschiede in Kommunikation und Sprache, soziale Interaktion, sowie wiederholende Verhaltensweisen und eingeschränkte Interessen sein. Die Symptome variieren stark zwischen betroffenen Personen und können im Verlauf des Lebens wechseln oder sich verändern.
Behandlungsmöglichkeiten zielen darauf ab, die Fähigkeiten zu fördern, Herausforderungen zu reduzieren und die Lebensqualität zu verbessern. Dazu gehören verhaltenstherapeutische Ansätze, Sprach- und Kommunikationstherapie, Ergotherapie, frühkindliche Förderprogramme sowie individuelle schulische Unterstützung. In vielen Fällen kommen auch medizinische Behandlungen zum Einsatz, um begleitende Probleme wie Schlafstörungen, Angst oder Epilepsie zu behandeln. Wichtige Prinzipien sind frühzeitige Diagnose, individuelle Anpassung der Unterstüzungsmaßnahmen und enge Zusammenarbeit mit Familien, Schulen und therapeutischen Fachpersonen.
Autismus tritt weltweit unterschiedlich häufig auf, Schätzungen zufolge betrifft es etwa 1 von 100 Kindern. Die Diagnosekriterien und Überwachungsmethoden können je nach Land variieren, daher liegen die Zahlen in Regionen oft etwas auseinander. Es gibt kein Heilmittel, aber viele Menschen mit ASS entwickeln im Laufe des Lebens Fähigkeiten, Kompetenzen und Strategien, um ein erfülltes und autonomes Leben zu führen.

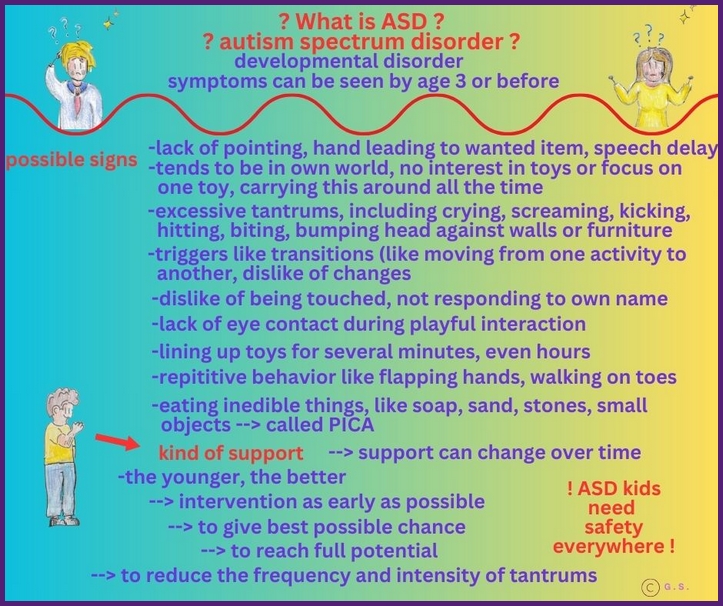
AI / KI in der Forschung
Der Einsatz von KI (Künstliche Intelligenz) nimmt rapide zu. KI bietet zwar spannende Möglichkeiten, um die Forschung voranzubringen, bringt aber auch gewisse Herausforderungen mit sich.
Was sind die wichtigsten Vorteile und welche Bedenken sollten wir beachten?
KI kann große Datenmengen schnell analysieren, Muster erkennen und Zusammenhänge aufdecken, die menschlichen Forschern oft verborgen bleiben. Sie unterstützt bei der Hypothesenbildung, der Optimierung von Experimenten und der automatisierten Datenerfassung, wodurch sich Zeit und Ressourcen sparen lassen. KI-gestützte Modelle ermöglichen bessere Vorhersagen.
In der Bild- und Signalverarbeitung liefert sie präzise Diagnosen oder Klassifikationen. KI kann außerdem repetitive Aufgaben automatisieren, die Reproduzierbarkeit erhöhen und skalierbare Analysen ermöglichen, die menschliche Kapazitäten übersteigen.
Mit KI verbunden sind Risiken wie Verzerrungen in den Daten, die zu fehlerhaften Vorhersagen führen können, sowie Transparenzprobleme bei komplexen Modellen. Abhängigkeit von großen, hochwertigen Datensätzen kann die Forschung verzerren, insbesondere wenn Daten unvollständig oder unfair erhoben werden. Es besteht die Gefahr, dass KI zu einer Übervertrauen in Modelle führt. Sicherheits- und Datenschutzaspekte müssen beachtet werden, besonders bei sensiblen Gesundheits- oder personenbezogenen Daten. Schließlich besteht das Risiko, dass KI menschliche Expertise und kritische Reflexion ersetzt statt ergänzt, was zu einem Verlust von Kontextwissen führen könnte.


Forschungsmodelle
"in vivo vs. in vitro"
Die biomedizinische Forschung stützt sich auf verschiedene Modelle, um Biologie, Krankheitsmechanismen und potenzielle Therapien zu untersuchen. In-vivo-Modelle beinhalten lebende Organismen und erfassen komplexe Wechselwirkungen innerhalb eines gesamten Systems, während In-vitro-Modelle isolierte Zellen, Gewebe oder Biomaterialien in einer kontrollierten Umgebung verwenden. Zusammen helfen diese Modelle Forschern, grundlegende Prozesse zu verstehen, Hypothesen zu testen und Erkenntnisse in klinische Fortschritte umzusetzen.
In-vivo- und In-vitro-Modelle sind zusammen mit ihren Stärken, Grenzen und typischen Anwendungen für die moderne Forschung von entscheidender Bedeutung.
In-vitro-Modelle sind Untersuchungen außerhalb eines lebenden Organismus, zum Beispiel Zellkulturen, Gewebeproben, Organoide oder Mikroorganismen. Es besteht eine größere Kontrolle über Umweltbedingungen, eine höhere Reproduzierbarkeit und ein geringeres ethisches Risiko im Vergleich zu tierischen oder systemischen Studien. Aber es fehlt oft die umfassende Repräsentativität ganzer Organismen, und komplexe Interaktionen können fehlen.
In-vivo-Modelle sind Experimente oder Beobachtungen, die innerhalb eines lebenden Organismus stattfinden, zum Beispiel in Tieren oder Menschen. Es können komplexe Interaktionen zwischen Organen, Immunsystem und Stoffwechsel berücksichtigt und direkt endogene Effekte und Krankheitsverläfe beobachtet werden.




Neuronale Erkrankungsmechanismen
Channelopathien, mTORopathien, Rasopathien und Enzephalopathien sind Begriffe aus der Neurologie/Genetik, die unterschiedliche Ursachen und Auswirkungen neuronaler Erkrankungen erklären.
Channelopathien bezeichnet man Erkrankungen, bei denen Defekte oder Fehlregulationen von Ionenkanälen (wie Natrium-, Kalium-, Calciumkanäle) zentrale Rolle spielen. Diese Störung verändert das Membranpotential der Nervenzellen und damit die Ausschüttung von Neurotransmittern sowie die Weiterleitung von Nervensignalen. Die Folge kann epileptische Anfälle, Bewegungsstörungen, Muskelzittern oder Schmerzempfindlichkeiten sein.
mTORopathien beziehen sich auf Erkrankungen, bei denen der mTOR-Signalweg dysreguliert ist. Der mTOR-Weg steuert Zellwachstum, Proteinsynthese und Synapsenbildung. Eine Überaktivität oder Fehlbalance kann zu vermehrtem Zellwachstum, veränderten neuronalen Verbindungen und gestörter Synapsenbildung führen. Klinisch äußern sich mTORopathien häufig als Epilepsie, Entwicklungsverzögerungen, Autismus-Spektrum-Störungen und gelegentlich Tumoren wie tuberöse Sklerose-Komplexe.
Rasopathien sind genetische Störungen, die den RAS/MAPK-Signalweg betreffen. Dieser Weg reguliert Zellteilung, Differenzierung und Wachstum. Durch Mutationen kommt es zu einer fehlerhaften Signalweiterleitung, was zu einer Reihe von körperlichen Merkmalen (wie Gesichtsanomalien, Hyperflexibilität, Hautveränderungen&rpat; und neurologischen Problemen führen kann. Im Gehirn kann die Rasopathie zu Entwicklungsverzögerungen, Lernschwierigkeiten, Epilepsien oder anderen neurologischen Symptomen beitragen.
Enzephalopathien sind ein Oberbegriff für Erkrankungen, die das Gehirn primär in seiner Funktion beeinträchtigen. Sie können durch Infektionen, Stoffwechselstörungen, Toxine, genetische Ursachen oder strukturelle Läsionen verursacht werden. Neurologisch zeigen sich häufig Bewusstseinsstörungen, kognitive Beeinträchtigungen, Sprache oder Motorik betroffen. Enzephalopathien sind kein einzelnes Krankheitsbild, sondern ein Sammelbegriff für verschiedenste Ursachen, deren zentrale Folge eine gestörte Gehirnfunktion ist.
Die Auswirkungen dieser Pathien überschneiden sich teils (etwa Epilepsie oder Entwicklungsstörungen), unterscheiden sich aber in der zugrundeliegenden Biologie und oft in der Therapieansprache: Channelopathien können gezielt mit Ionkanalblockern behandelt werden, mTORopathien mit Medikamenten, die den mTOR-Weg modulieren, Rasopathien erfordern oft ein multifaktorielles, symptombezogenes Management, während Enzephalopathien je nach Ursache unterschiedlich therapiert werden müssen.


Sequenzierungstechnologien
Es gibt verschiedene Sequenzierungstechnologien. Bei der Short-Read-Sequenzierung werden kleine DNA-Stücke erzeugt, die sehr genau und kostengünstig sind, jedoch Probleme mit langen, sich wiederholenden Regionen und der Zusammenstellung vollständiger Transkripte haben.
Bei der Long-Read-Sequenzierung werden viel längere DNA- oder RNA-Fragmente gelesen, wodurch es einfacher ist, Transkripte in voller Länge und große Variationen zu erkennen, jedoch sind die Kosten in der Regel höher und die Fehlerraten größer (obwohl die Genauigkeit mit neueren Methoden sehr gut sein kann).
Read-Through bezieht sich auf die Transkription oder Translation, die über ein normales Stopp-Signal hinaus fortgesetzt wird. Read-Through-Sequenzierung untersucht das translatorische Read-Through – die Situation, in der ein Ribosom ein normales Stoppkodon ignoriert und die Translation in die nachgeschaltete Sequenz fortsetzt.
Forscher verwenden sequenzierungsbasierte Methoden, um dieses Phänomen zu untersuchen. Ribosom-Profiling (Ribo-seq) steht dabei im Vordergrund, da es ribosomgeschützte mRNA-Fragmente sequenziert und Aufschluss darüber gibt, ob Ribosomen an Stoppkodons pausieren oder diese passieren, was auf Read-Through hindeutet. Translationales Read-Through bedeutet, dass Ribosomen ein Stoppkodon überlesen und eine verlängerte Proteinvariante produzieren.
Wenn die STXBP1-mRNA Kontexte enthält, die das Überlesen von Stoppkodons begünstigen, könnten dadurch C-terminal verlängerte STXBP1-Proteinisoformen mit veränderter Funktion, Lokalisierung oder Interaktionen entstehen, was die Freisetzung synaptischer Vesikel beeinflussen könnte. In der STXBP1-Forschung könnte Read-Through die Rolle von STXBP1 bei der Bildung des SNARE-Komplexes und der Vesikelfreisetzung wesentlich beeinflussen. Das Überschreiten eines Stoppkodons durch Translation könnte theoretisch eine verlängerte STXBP1-Proteinvariante mit unterschiedlichen Funktionen und Interaktionen erzeugen.
Arigin-Read-Through ist eine Form des translatorischen Read-Through, bei der das Ribosom ein Stoppkodon ignoriert und die Translation in die nachgeschaltete Sequenz fortsetzt, wobei Überschüsse oder ungewöhnliche Codon-Kontexte am Stoppkodon es ermöglichen, Arginin als extragenitales Überschreiten zu integrieren. Das kann dann zu einer verlängerten STXBP1-Proteinvariante mit veränderter Funktion oder Interaktion führen.


Induzierte pluripotente Stammzellen (iPSCs)
Induzierte pluripotente Stammzellen (iPSCs) sind adulte Zellen, die in einen pluripotenten Zustand zurückprogrammiert wurden und sich in viele Zelltypen differenzieren können. Sie werden in der Regel durch die Einführung einer Reihe von Transkriptionsfaktoren (z. B. Oct4, Sox2, Klf4 und c-Myc) in somatische Zellen wie Hautfibroblasten oder Blutzellen unter Verwendung von Methoden wie integrierenden oder nicht integrierenden viralen Vektoren oder episomalen Plasmiden erzeugt. iPSCs bieten eine patienten- oder isogene Linienplattform, die den genetischen Kontext erfasst und die Differenzierung in relevante Nervenzelltypen ermöglicht.
Induzierte pluripotente Stammzellen (iPSCs) bieten eine leistungsstarke Plattform für die Untersuchung der STXBP1-bezogenen Biologie und Erkrankungen. iPSCs ermöglichen Patienten- und isogene Linienmodelle, die den genetischen Kontext erfassen und eine Differenzierung in relevante Nervenzelltypen ermöglichen, wodurch ein für den Menschen relevantes System zur Untersuchung der STXBP1-Funktion bei der synaptischen Übertragung, der neuronalen Entwicklung und der Netzwerkaktivität bereitgestellt wird. Mithilfe von iPSCs können Forscher STXBP1-assoziierte Erkrankungen modellieren, potenzielle therapeutische Strategien untersuchen und erforschen, wie genetische Varianten oder Regulationsmechanismen die STXBP1-Expression und die Dynamik der Vesikelfreisetzung beeinflussen, mit dem Potenzial, die Ergebnisse in gezielte Interventionen umzusetzen.


All diese RNAs – die Bedeutung von miRNAs
Es gibt viele Arten von RNAs, die jeweils unterschiedliche Funktionen erfüllen. miRNAs sind wichtig, da sie die Genexpression regulieren, indem sie an Ziel-mRNAs binden und deren Abbau oder Translationshemmung bewirken. Sie beeinflussen eine Vielzahl biologischer Prozesse, darunter Entwicklung, Zelldifferenzierung, Proliferation, Apoptose und Reaktionen auf zellulären Stress.
Es ist möglich, Kombinationsstrategien mit miRNA-Inhibitoren oder -Mimetika einzusetzen, um STXBP1-Signalwege zu modulieren und die den synaptischen Phänotypen zugrunde liegenden Signalwege zu interpretieren. miRNAs werden in der ASO-Forschung im Hinblick auf Off-Target-Effekte berücksichtigt.
Da STXBP1 ein Regulator der synaptischen Freisetzung ist, könnte die gezielte Beeinflussung seiner mRNA mit ASOs die Freisetzung von Neurotransmittern beeinflussen.
Studien zufolge können konditionale ASOs mit jeder miRNA individuell gestaltet werden, um die ASO-Aktivierung in Zielzellen zu steuern und gleichzeitig unerwünschte Effekte in Nicht-Zielzellen zu reduzieren.



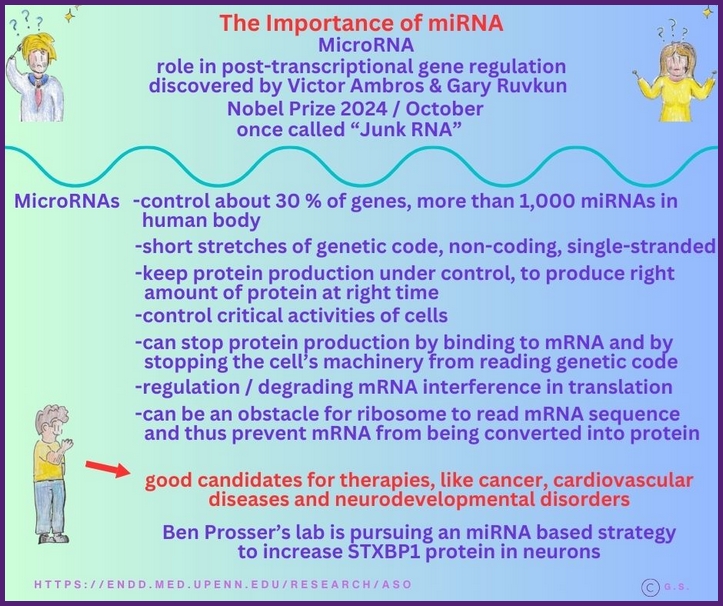
Genotyp vs. Phänotyp
Bei seltenen Krankheiten bestimmt der Genotyp einer Person – also ihre genetische Veranlagung – nicht immer direkt ihren Phänotyp, also die beobachtbaren Merkmale oder Symptome. Personen mit derselben Mutation können unterschiedliche Symptome aufweisen, und ähnliche Symptome können unterschiedliche genetische Ursachen haben. Umweltfaktoren und andere genetische Einflüsse beeinflussen häufig die Expression von Genen, wodurch die Beziehung zwischen Genotyp und Phänotyp komplex und nicht immer eindeutig ist.
Das Erkennen dieser fehlenden Eins-zu-Eins-Beziehung ist entscheidend für eine genaue Diagnose und personalisierte Behandlungsansätze bei seltenen Krankheiten.
Der Genotyp bei STXBP1 beschreibt die genetische Ausstattung des Individuums, also die spezifischen Varianten im STXBP1-Gen, wie Mutationen oder SNPs. Er gibt das Potenzial auf DNA-Ebene wieder, während der Phänotyp die beobachtbaren Merkmale umfasst, die aus dem Zusammenspiel von Genetik, Umwelt und weiteren Faktoren resultieren. Bei STXBP1 schließen Phänotypen typischerweise früh einsetzende Epilepsie, Entwicklungsverz/ouml;gerung, kognitive Beeinträchtigungen und andere neurologische Merkmale ein. Es ist wichtig zu beachten, dass der Genotyp nicht zwingend mit dem Phänotyp übereinstimmt oder korreliert.
In der Forschung ist es essenziell, Genotyp und Phänotyp nicht einfach zu verwechseln oder zu vermischen, da Unterschiede im Genotyp, etwa LOF versus GOF Mutationen, zu unterschiedlichen klinischen Bildformen beitragen, unabhängig davon, ob eine direkte Korrelation besteht. Die Unterscheidung hilft, Prognose und Therapiemöglichkeiten zu verfeinern und unterstützt die Abgrenzung zwischen genetischer Ursache und beobachteten Merkmalen. Im Design von Studien verhindert eine klare Trennung oder bewusste Verknüpfung von Genotyp und Phänotyp Missverständnisse und erleichtert die Interpretation der Ergebnisse, egal ob ein genotype-first oder phenotype-first Ansatz verfolgt wird.


LOF vs. GOF
Im Zusammenhang mit der Genetik, die sich auf Mutationen in Genen konzentriert, haben LOF und GOF spezifische Bedeutungen.
LOF (Loss of Function, Funktionsverlust) bezieht sich auf Mutationen, die die Aktivität des Genprodukts verringern oder aufheben, was häufig zu einer verminderten oder fehlenden Proteinfunktion führt.
GOF (Gain of Function, Funktionsgewinn) bezieht sich auf Mutationen, die die Aktivität des Genprodukts verstärken, neuartig gestalten oder auf andere Weise verändern und damit Effekte hervorrufen, die im normalen Allel nicht vorhanden sind.
Beide Konzepte sind von zentraler Bedeutung für das Verständnis von Krankheitsmechanismen, Genotyp-Phänotyp-Beziehungen und therapeutischen Strategien.
In der Forschung beschreiben LOF und GOF die funktionellen Auswirkungen von Varianten, die Aufschluss über die Pathogenitätsbewertung, Erwartungen hinsichtlich klinischer Phänotypen und die Interpretation von Versuchsergebnissen geben. Die Unterscheidung zwischen LOF und GOF hilft bei der Priorisierung von Varianten für die funktionelle Validierung und bei der Konzeption von Experimenten zur Überprüfung mechanistischer Hypothesen.
LOF (Loss of Function, Funktionsverlust) in STXBP1 bedeutet, dass Mutationen die Funktion des STXBP1-Proteins reduzieren oder aufheben und dadurch die Freisetzung synaptischer Vesikel und die neuronale Kommunikation beeinträchtigen. Dies wird häufig mit früh einsetzender Epilepsie, geistiger Behinderung und Entwicklungsverzögerungen in Verbindung gebracht. Typische Mutationstypen sind Nonsense-Mutationen, Frameshift-Mutationen, kritische Spleißstellenveränderungen oder Deletionen, die die Proteinproduktion oder essentielle Domänen stören. Geben Sie bei der Berichterstattung "Loss-of-Function-Varianten von STXBP1" an, sofern funktionelle oder biochemische Belege dafür vorliegen.
GOF (Gain of Function) in STXBP1 bedeutet, dass Mutationen eine erhöhte oder neue STXBP1-Aktivität oder eine dysregulierte Regulation der synaptischen Vesikelfreisetzung bewirken. Dies ist weniger häufig, wurde jedoch berichtet und kann zu Übererregbarkeit oder atypischen Phänotypen führen.

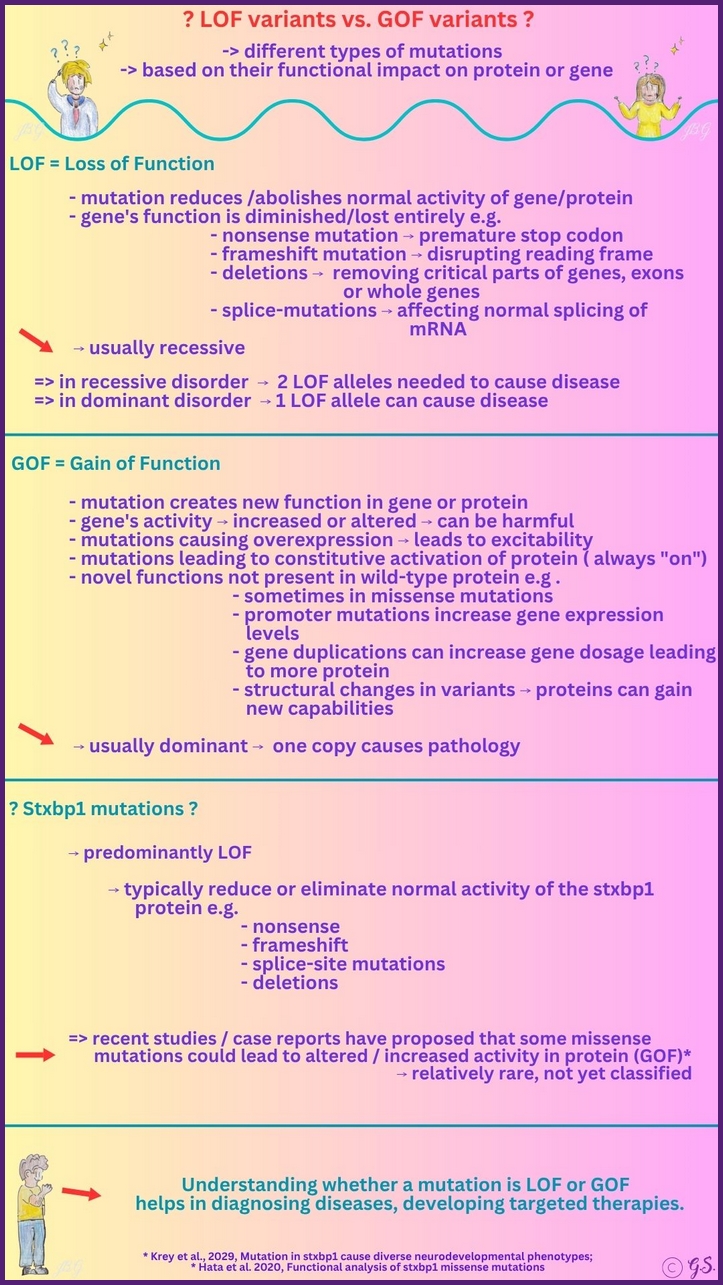
MRT vs. ULF-MRT
Die Bildgebung von Epilepsie mit Ultra-Niedrigfeld-MRT (Ultra-Low-Field MRI)
ist ein neues Forschungsfeld, das die detaillierte Darstellung von Hirnstrukturen verbessern könnte.
Ultra-Low-Field-MRT könnte mit seinen speziellen Spulentechniken potenziell noch mehr Details sichtbar machen. Die Technik wird derzeit erforscht, um die Diagnose, insbesondere bei schwer behandelbaren Epilepsien, zu verbessern und die prächirurgische Planung zu präzisieren.
Die Magnetresonanztomographie (MRT) ist ein medizinisches Bildgebungsverfahren, bei dem mithilfe starker Magnetfelder und Radiowellen hochauflösende Bilder von Weichteilen erzeugt werden. Sie eignet sich hervorragend für die detaillierte Darstellung des Gehirns, des Rückenmarks, der Gelenke und der Bauchorgane und ist damit ein wichtiger Bestandteil für eine genaue Diagnose und Behandlungsplanung. Die Ultra-Low-Field-MRT (ULF-MRT) hingegen basiert auf wesentlich schwächeren Magnetfeldern und zeichnet sich durch kleinere, kostengünstigere Systeme aus, die einfacher zu warten und oft auch tragbar sind. Während die ULF-MRT in der Regel eine geringere Bildqualität und längere Scanzeiten als die herkömmliche MRT bietet, hat sie Vorteile in Bezug auf Zugänglichkeit, geringere Kosten und Flexibilität beim Einsatz, sodass Bildgebung in ressourcenbeschränkten Umgebungen, am Krankenbett oder an abgelegenen Orten möglich ist.
Kurz gesagt, die MRT ist führend in Bezug auf Bildqualität und Diagnosemöglichkeiten, während die ULF-MRI Kosten, Tragbarkeit und Anpassungsfähigkeit in den Vordergrund stellt – jede Technik erfüllt unterschiedliche klinische und Forschungsanforderungen.
Jüngste Forschungsergebnisse deuten jedoch darauf hin, dass sich die Bildqualität, Zuverlässigkeit und klinische Verwendbarkeit der Ultra-Low-Field-MRT (ULF-MRT) verbessert.
Fortschritte bei der Hardware, wie verbesserte Sensoren und leistungsfähigere Elektronik, sowie Softwareentwicklungen in den Bereichen Bildrekonstruktion und Rauschunterdrückung verringern den Abstand zur herkömmlichen MRT. Zwar liegt die ULF-MRT in Bezug auf Auflösung und Scangeschwindigkeit im Allgemeinen noch immer zurück, doch diese Verbesserungen erweitern ihr Potenzial für die Bildgebung am Krankenbett, die Point-of-Care-Diagnostik und den Einsatz in ressourcenbeschränkten Umgebungen.
Insgesamt entwickeln sich sowohl die MRT als auch die ULF-MRT weiter, wobei die ULF-MRT immer wettbewerbsfähiger wird, da die Forschung in praktische, einsetzbare Systeme umgesetzt wird.


Modellorganismen in der Forschung
Modellorganismen sind nicht-menschliche Spezies, die zum Verständnis biologischer Prozesse verwendet werden, die auch beim Menschen vorkommen. Sie bieten praktische Vorteile wie kurze Generationszeiten, einfache Pflege, gut annotierte Genome und umfangreiche Werkzeuge. Sie geben Aufschluss über grundlegende zelluläre und entwicklungsbiologische Mechanismen und ermöglichen eine robuste genetische und umweltbezogene Manipulation, wodurch sie Erkenntnisse über die menschliche Gesundheit und Krankheiten liefern. Auch ethische und logistische Überlegungen sprechen für ihre Verwendung.
Es gibt jedoch Einschränkungen, da die Ergebnisse möglicherweise nicht direkt auf den Menschen übertragbar sind; für einige menschliche Merkmale sind Säugetiermodelle erforderlich. Die Validierung über Speziesgrenzen hinweg ist wichtig.


Mitochondriale Erkrankungen
Mitochondriale Erkrankungen sind eine Untergruppe genetischer Erkrankungen, die durch Probleme in den Mitochondrien, den energieproduzierenden Teilen unserer Zellen, verursacht werden. Sie können durch Mutationen in der mitochondrialen DNA, die ausschließlich von der Mutter vererbt wird, oder durch Mutationen in Kerngenen, die die Mitochondrienfunktion beeinträchtigen, entstehen. Dieser doppelte genetische Ursprung bedeutet, dass mitochondriale Erkrankungen mit einer Vielzahl von Symptomen und Organbeteiligung einhergehen können, die häufig Gewebe mit hohem Energiebedarf wie Gehirn, Muskeln, Herz und Augen betreffen.
Im Gegensatz dazu sind breiter gefasste genetische Störungen solche, die durch Variationen in Genen verursacht werden, die überall im Genom auftreten können, nicht nur im mitochondrialen Genom, und sie können verschiedenen Vererbungsmustern folgen, darunter autosomal-dominante, autosomal-rezessive, X-chromosomale oder De-novo-Mutationen.
Während genetische Störungen auch in ihren klinischen Manifestationen variieren, zeichnen sich mitochondriale Störungen durch ihre Ursache in der Mitochondrienbiologie und ihre charakteristischen Muster von Energieversagen aus, die manchmal mit Laktatazidose, Belastungsintoleranz und Beteiligung mehrerer Systeme einhergehen. Das Verständnis dieses Unterschieds hilft bei der Anpassung diagnostischer Ansätze, einschließlich gezielter Gentests und Bewertungen der Mitochondrienfunktion, sowie von Behandlungsstrategien, die sich auf Energieoptimierung, unterstützende Therapien und, sofern verfügbar, die Teilnahme an klinischen Studien konzentrieren.
Genom vs. Pangenom
Unsere genetische Ausstattung, das Genom, enthält alle Informationen, die unseren Körper und seine Funktionen bestimmen. Innerhalb einer Spezies gibt es jedoch viele Unterschiede zwischen den Genomen einzelner Organismen. Das Pangenom fasst diese gesamte genetische Vielfalt zusammen und zeigt, welche Gene in einer Population vorhanden sind und welche Variationen auftreten können.
Das Verständnis des Unterschieds zwischen einem Genom und einem Pangenom ist entscheidend, um zu verstehen, wie die genetische Vielfalt innerhalb einer Spezies untersucht wird. Ein Genom bezeichnet den vollständigen Satz an genetischem Material, der in einem einzelnen Individuum oder Organismus vorhanden ist, einschließlich aller Gene und nichtkodierenden Regionen. Es liefert den Bauplan für die Biologie dieses Organismus, erfasst jedoch nur eine Momentaufnahme des genetischen Inhalts der Art zu einem bestimmten Zeitpunkt.
Im Gegensatz dazu umfasst ein Pangenom die vollständige Komplementärgruppe aller Gene und genetischen Elemente, die bei allen Individuen einer Art zu finden sind. Es umfasst das Kerngenom, das allen Individuen gemeinsam ist, sowie das akzessorische oder entbehrliche Genom, das sich zwischen den Individuen unterscheidet und zu Unterschieden in Merkmalen, Anpassungen und Reaktionen auf die Umwelt beitragen kann. Die Betrachtung der Genomik im Hinblick auf ein Pangenom erkennt an, dass kein einzelnes Referenzgenom die gesamte genetische Vielfalt einer Spezies erfassen kann, und unterstreicht die dynamische, sich entwickelnde Natur von Genomen, je mehr Individuen sequenziert werden. Dieser Wandel von einer einzigen, statischen Referenz hin zu einem umfassenderen Pangenomkonzept hat tiefgreifende Auswirkungen auf das Verständnis von Evolution, Ökologie und Medizin und ermöglicht genauere Genomassemblierungen, eine bessere Interpretation genetischer Variationen und effektivere Strategien für Züchtung, Erhaltung und personalisierte Therapien.


Klinische Forschung vs. medizinische Forschung
Klinische Forschung und medizinische Forschung sind verwandte, aber unterschiedliche Konzepte, die beide für die Verbesserung der Patientenversorgung und den wissenschaftlichen Fortschritt von entscheidender Bedeutung sind.
Die klinische Forschung konzentriert sich darauf, zu untersuchen, wie Krankheiten Menschen beeinflussen und wie Behandlungen bei echten Patienten wirken. Dazu gehört die Entwicklung und Erprobung neuer Therapien, Medikamente, medizinischer Geräte und Verfahren im Rahmen von Studien, an denen Teilnehmer beteiligt sind. Ziel ist es, die Sicherheit, Wirksamkeit und den Nutzen in realen medizinischen Umgebungen zu ermitteln, damit Ärzte fundierte Entscheidungen über die Behandlung von Patienten treffen können.
Medizinische Forschung hingegen ist ein weiter gefasster Begriff, der alle wissenschaftlichen Untersuchungen umfasst, die darauf abzielen, Gesundheit, Krankheit und die Biologie des menschlichen Körpers zu verstehen. Sie umfasst Grundlagenforschung in Laboren, Tierversuche, genetische und translationale Studien sowie klinische Studien. Die medizinische Forschung versucht, Krankheitsmechanismen aufzudecken, neue Ansatzpunkte für Interventionen zu identifizieren und Erkenntnisse aus dem Labor in praktische Anwendungen für die Patientenversorgung umzusetzen.
Beide Arten der Forschung sind wichtig, da sie sich gegenseitig ergänzen. Die medizinische Forschung schafft das grundlegende Wissen, das erklärt, wie und warum Krankheiten auftreten und wie neue Interventionen wirken könnten. Die klinische Forschung wendet dieses Wissen auf den Menschen an, testet, was in realen klinischen Umgebungen funktioniert, und bewertet die Sicherheit und Wirksamkeit bei verschiedenen Patientengruppen. Zusammen treiben sie die Entwicklung besserer Diagnostik, Behandlungen und Präventionsstrategien voran und verbessern letztendlich die Gesundheitsergebnisse und die evidenzbasierte Praxis.


Numts und Numtogenese
Numts sind Teile der mitochondrialen DNA, die im Laufe der Zeit in den Zellkern gelangt sind. Numtogenese beschreibt, wie diese mitochondrialen Teile im Zellkern entstehen und sich dort ansammeln, und welche Bedeutung sie für Genome und die Evolution haben.
Numts sind die tatsächlichen mitochondrialen Fragmente, die sich nun im Zellkern befinden. Numtogenese ist der Prozess, durch den diese Fragmente im Genom erscheinen und sich dort ausbreiten. Numts werden untersucht, um mehr über Genome und das Lesen von DNA-Stammbäumen zu erfahren. Die Numtogenese wird untersucht, um zu verstehen, wie sich DNA im Laufe der Zeit bewegt und verändert.
Das Wissen über Numtogenese hilft uns, bessere Stammbäume des Lebens zu erstellen. Es hilft auch in der Forensik und Medizin, indem es echte mtDNA von Numts unterscheidet. Es zeigt, wie sich Genome verändern und wachsen.
Es ist leicht, mtDNA mit Numts zu verwechseln. Numts können zu Fehlern bei DNA-Tests führen. Viele Numts haben möglicherweise keine Funktion, sondern befinden sich einfach nur im Genom. Numts kommen in vielen Geweben vor, darunter auch im Gehirn. Mitochondriale DNA-Fragmente können in verschiedenen Zelltypen in das Kerngenom integriert werden. Das Gehirn ist in dieser Hinsicht nichts Besonderes, aber die Erkennung von Numts kann bei der Sequenzierung schwierig sein, da das Gehirngewebe viele Mitochondrien und eine hohe Komplexität aufweist.


Gentherapie, Genbearbeitung und Zelltherapie
Gentherapie, Genbearbeitung und Zelltherapie sind sich überschneidende, aber dennoch unterschiedliche Ansätze und Technologien bzw. Anwendungsfelder an der Spitze der modernen Medizin, die jeweils darauf abzielen, Krankheiten zu behandeln oder zu verhindern, indem sie auf biologische Mechanismen auf molekularer und zellulärer Ebene abzielen.
Gentherapie bezieht sich im Allgemeinen auf Strategien, bei denen genetisches Material in die Zellen eines Patienten eingeführt, ersetzt oder stillgelegt wird, um eine therapeutische Wirkung zu erzielen. Gentherapie zielt darauf ab, genetische Informationen direkt in Zellen zu verändern, um Krankheiten zu behandeln. Dabei wird entweder funktionelles Genmaterial geliefert, um ein defektes oder fehlendes Gen zu ersetzen, oder schädliche genetische Sequenzen werden deaktiviert. Häufig erfolgen die Anwendungen durch Trägervektoren wie Adeno-assoziierte Viren (AAV) oder Lipid-Nanopartikel, die die gewünschte DNA oder RNA in die Zielzellen transportieren. Gentherapie kann persistente, dauerhafte Veränderungen bewirken und wird vor allem bei genetischen Erkrankungen erforscht, darunter bestimmte Bluterkrankungen, metabolische Störungen oder neurodegenerative Erkrankungen. Wichtige Aspekte sind Sicherheit, Immunantworten, Gewebe-Spezifität und Langzeitüberwachung.
Geneditierung umfasst dagegen präzise, gezielte Veränderungen der bestehenden DNA im Genom selbst, wodurch krankheitsverursachende Mutationen korrigiert, pathogene Sequenzen unterbrochen oder vorteilhafte genetische Veränderungen mit hoher Spezifität eingeführt werden können. Technologien wie CRISPR-basierte Systeme, Base Editoren und Prime Editoren sind Beispiele f&uum;r diesen Bereich. Ziel ist es, Gene mutativ zu verändern, Funktions- oder Expressionsmuster zu korrigieren oder Modelle für die Forschung zu schaffen. Im Gegensatz zur klassischen Gentherapie verändert die Genbearbeitung direkt die genetische Sequenz an ihrem ursprünglichen Ort, was potenziell dauerhafte Effekte haben kann. Herausforderungen umfassen Off-Target-Effekte, Liefermethoden, ethische Implikationen und regulatorische Rahmenbedingungen.
Die Zelltherapie konzentriert sich auf die Verabreichung lebender Zellen zur Reparatur, zum Ersatz oder zur Unterstützung erkrankter Gewebe. Bei der Zelltherapie wird neues Zellmaterial eingeführt, das funktionelle Vorteile bieten und therapeutische Ergebnisse erzielen kann. Typische Beispiele sind Stammzelltherapien zur Regeneration von Knochenmark oder Gewebe, Immunzelltherapien wie CAR-T-Zelltherapie gegen Krebs oder die Transplantation von MPCs, neuralen Progenitorzellen oder mesenchymalen Stammzellen zur Behandlung von neurodegenerativen oder entzündlichen Erkrankungen. Bei der Zelltherapie spielt die Herkunft der Zellen, ihre Differenzierung, das Transplantationsziel und die Sicherheit eine zentrale Rolle. Wichtige Themen sind Immunverträglichkeit, Kontamination, Kontrolle der Zelldynamik und Langzeitwirkung.
Die Bedeutung dieser Ansätze liegt in ihrem Potenzial, Krankheiten mit begrenzten oder keinen wirksamen Behandlungsmöglichkeiten anzugehen und personalisierte und dauerhafte Interventionen anzubieten, die auf die Ursachen abzielen und nicht nur die Symptome behandeln.
Alle drei Ansätze stoßen auf gemeinsame ethische, regulatorische und sicherheitstechnische Fragestellungen, etwa zu Langzeitfolgen, Transferrisiken, Replikation von Veränderungen und dem Zugang zu Therapien. Je nach Krankheit, Verfügbarkeit von Delivery-Systemen, Vertrauen in die Evidenz und regulatorischen Vorgaben kann eines oder eine Kombination dieser Ansätze sinnvoll sein.

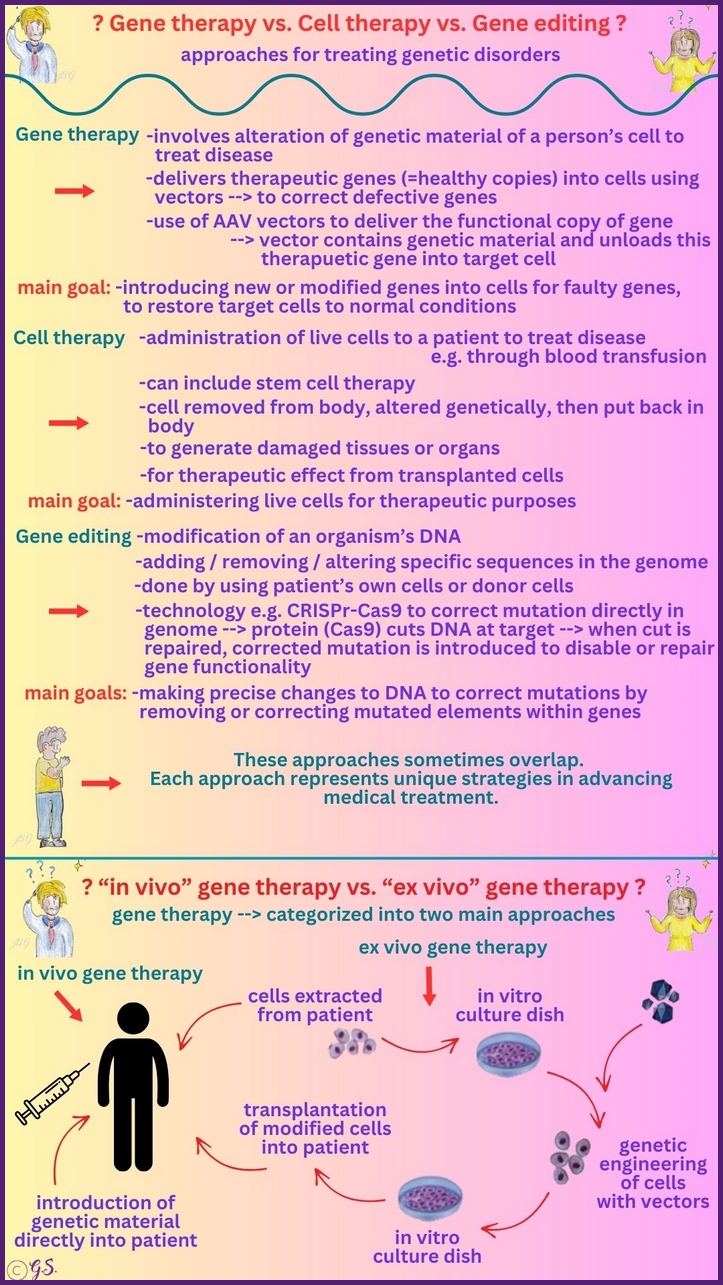
Klinisches-EEG vs. Forschung-EEG
Die Elektroenzephalographie (EEG) ist ein wichtiges Instrument in der Neurologie zur Aufzeichnung der elektrischen Aktivität des Gehirns und für die Beurteilung von Epilepsie unverzichtbar. Im Laufe der Jahre haben sich die EEG-Techniken von kurzen Standardaufzeichnungen zu längeren, multimodalen Ansätzen entwickelt, darunter erweiterte Überwachung und Videointegration sowie spezielle Methoden während des Schlafs oder unter provokativen Bedingungen. Diese vielfältigen Formen helfen Ärzten dabei, epileptiforme Aktivitäten zu erkennen, Anfälle zu klassifizieren, epileptogene Zonen zu lokalisieren und das Ansprechen auf Behandlungen zu überwachen. Obwohl das EEG von unschätzbarem Wert ist, ist es nur ein Teil des diagnostischen Puzzles und sollte im Zusammenhang mit der Krankengeschichte und anderen Untersuchungen interpretiert werden. Heute wird das EEG mit Bildgebung, Neuropsychologie und Genetik kombiniert, und automatisierte Analysen sowie maschinelles Lernen unterstützen zunehmend die Interpretation.
Biomarker spielen eine wesentliche Rolle im Zusammenhang mit EEG-Diagnostik, sowohl klinisch als auch in der Forschung.
Klinisch: EEG-Biomarker wie bestimmte Muster (z. B. epileptiforme Entladungen, späte Aktivität nach Reizung, Burst-Suppression) oder Quantitätskennzahlen aus dem EEG (z. B. Hintergrundaktivität, Latency- und Amplitudenveränderungen(unterstützen die Diagnosestellung, Verlaufskontrolle und Einschätzung der Therapieresponse. In manchen Situationen helfen EEG-basierte Biomarker, Prognosen zu erstellen (z. B. Risiko für Anfallfreiheit unter Medikation) oder Unterschiede zwischen Epilepsie-Syndromen zu differenzieren.
Forschungsseitig: Biomarker-Analysen im EEG umfassen fortgeschrittene Messgrößen wie Power Spectral Density, funktionelle Konnektivität, Trendanalysen über Langzeit-EEG, Event-Related Potentials (ERPs) und Muster, die mit kognitiven Prozessen oder Epileptogenese assoziiert sind. Ziel ist oft, neue diagnostische Marker, Frühwarnsignale, oder Endpunkte für Therapieversuche zu identifizieren.


Prime Editing vs. Enzymersatztherapie
Prime Editing und Enzymersatztherapie sind zwei unterschiedliche Ansätze zur Behandlung genetischer Erkrankungen, die jeweils auf unterschiedlichen wissenschaftlichen Paradigmen basieren und darauf abzielen, die normale Zellfunktion wiederherzustellen. Sie kommen in der Medizin und Biotechnologie zum Einsatz.
Prime Editing ist eine Genom-Editierungstechnik der nächsten Generation, bei der neue genetische Informationen direkt an eine bestimmte Stelle in der DNA geschrieben werden, wodurch krankheitsverursachende Mutationen mit programmierbarer Präzision an ihrer Quelle korrigiert werden können. Prime Editing verändert also die DNA-Sequenz an bestimmten Genomstellen direkt. Sie kombiniert eine katalytisch beeinträchtigte CRISPR-Cas9-Nickase mit einer reversen Transkriptase und einer Prime-Editing-Guide-RNA, um gewünschte genetische Veränderungen zu schreiben, ohne Doppelstrangbrüche zu erzeugen. Dies ermöglicht präzise Substitutionen, Insertionen oder Deletionen und wird für genetische Veränderungen in Zellen oder Organismen verwendet, wobei Überlegungen hinsichtlich Spezifität, Verabreichung, Off-Target-Effekten und ethischen/regulatorischen Richtlinien zu berücksichtigen sind.
Im Gegensatz dazu behandelt die Enzymersatztherapie Krankheiten auf biochemischer Ebene, indem sie funktionelle Enzyme bereitstellt, um defekte oder fehlerhafte Enzyme zu kompensieren und so pathologische Prozesse zu lindern, ohne den zugrunde liegenden genetischen Code zu verändern. Der Schwerpunkt liegt auf der Verwendung natürlicher oder künstlich hergestellter Enzyme, die auf Substrate im Körper einwirken, wobei der Fokus auf der Verabreichung, der Zielgenauigkeit, der Sicherheit und der Überwachung liegt.
Zusammen veranschaulichen diese Strategien das Spektrum therapeutischer Innovationen – die eine zielt darauf ab, das Genom umzuschreiben, um vererbte Defekte zu korrigieren, während die andere versucht, die biochemischen Defizite zu beheben, die das Fortschreiten der Krankheit vorantreiben. Dies zeigt, wie sich durch komplementäre Perspektiven die Möglichkeiten für Patienten und Forscher erweitern lassen, während wir nach sichereren und wirksameren Behandlungsmethoden suchen.
Prime Editing System


Unser Gehirn - nur ein Computersystem?
Das Gehirn wird oft mit einem Computer verglichen, aber es leistet mehr als jede Maschine.
Das Gehirn ist kein Computersystem, da es Informationen flexibel und parallel verarbeitet; es lernt, passt sich an und reguliert unzählige Systeme in Echtzeit. Es verschaltet sich neu, verarbeitet Emotionen und verbindet Biologie mit dem menschlichen Verhalten.
Computer folgen feste Regeln, basieren auf vordefinierten Architekturen und ihre Struktur wird durch Lernen nicht grundlegend verändert.
Das Gehirn integriert Wahrnehmung, Gedächtnis und Handlung auf ganzheitliche, kontextabhängige Weise, toleriert Störungen und Verletzungen und erzeugt bewusste Erfahrungen, was keine Eigenschaften aktueller Computersysteme sind.
Kurz gesagt, das Gehirn ist ein biologisches, plastisches, energieeffizientes System, das durch Veränderung seiner Verknüpfungen lernt, während Computer deterministische oder probabilistische Maschinen mit festen Architekturen und vordefinierter Logik sind.
Eine wichtige Kontrollinstanz in diesem Gehirn-System ist die Blut-Hirn-Schranke (BHS), die das Nervengewebe schützt und gleichzeitig kontrolliert, was ins Gehirn gelangt und wie es dessen Funktion beeinflusst. Es handelt sich um eine selektive Barriere aus Endothelzellen, die nur wichtige Nährstoffe durchlässt.
Die Blut-Hirn-Schranke fügt eine wichtige Ebene hinzu – sie entscheidet, welche Chemikalien ins Gehirn gelangen dürfen, und bestimmt, wie Signale interpretiert und Reaktionen erzeugt werden. Die BHS ist ein entscheidender Gatekeeper in diesem System, der dafür sorgt, dass die neuronale Verarbeitung stabil und effizient bleibt. Keine echte, tatsächlich vorhandene Computerleistung oder Computerfunktion!



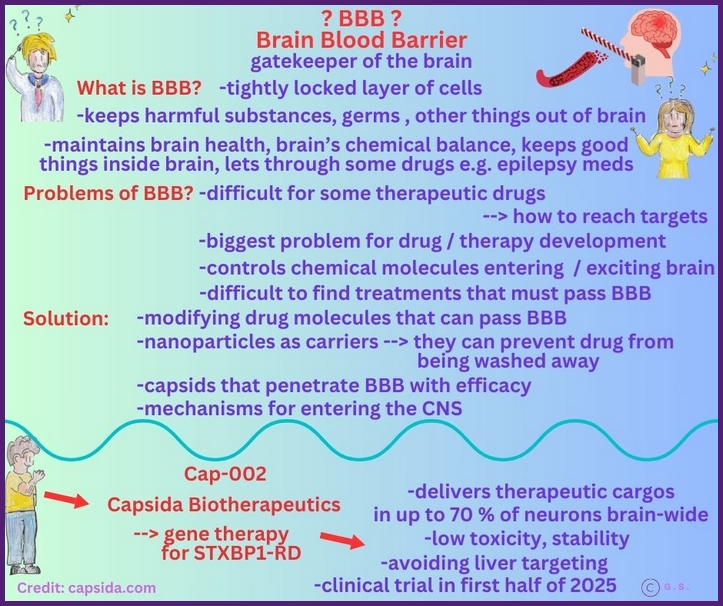
Genomsequenzierung
Die Genomsequenzierung ermöglicht es uns, den vollständigen DNA-Bauplan von Lebewesen zu entschlüsseln. Das Referenzgenom dient als Standard, als umfassende Karte, die die typische genetische Ausstattung einer Spezies darstellt. Im Gegensatz dazu offenbart eine individuelle Genomsequenz einzigartige genetische Variationen, die einen Menschen von einem anderen unterscheiden. Der Vergleich dieser beiden Sequenzen hilft uns, die genetische Vielfalt zu verstehen, Mutationen zu identifizieren, die mit Krankheiten in Verbindung stehen, und die personalisierte Medizin voranzubringen.
Die Sequenzierung von STXBP1 bezieht sich auf die Analyse des STXBP1-Gens, das für das Syntaxin-bindende Protein 1 kodiert, einen wichtigen Regulator der Neurotransmitterfreisetzung in Neuronen. Dieses Gen steht im Zusammenhang mit einer Reihe von neurologischen Entwicklungsstörungen und epileptischen Erkrankungen, darunter frühkindliche epileptische Enzephalopathie und andere Anfallsleiden. Durch die Sequenzierung können pathogene Varianten identifiziert werden – wie Missense-, Nonsense-, Frameshift-, Spleißstellenmutationen oder Veränderungen der Kopienzahl –, die die Funktion von STXBP1 stören und zur Erkrankung beitragen.
Sequenzierung ist notwendig, denn STXBP1-bedingte Erkrankungen weisen eine erhebliche klinische und genetische Heterogenität auf, und viele Patienten haben keine eindeutige Familienanamnese. Eine umfassende Sequenzierung hilft dabei, eine präzise genetische Diagnose zu stellen, die Prognose zu bestimmen, die Behandlung zu steuern, eine angemessene genetische Beratung zu ermöglichen und die Eignung für gezielte/experimentelle Therapien oder klinische Studien zu beurteilen. Sie kann auch zufällige Befunde mit weiterreichenden gesundheitlichen Auswirkungen aufdecken, was die Notwendigkeit einer sorgfältigen genetischen Beratung und Einwilligung unterstreicht.



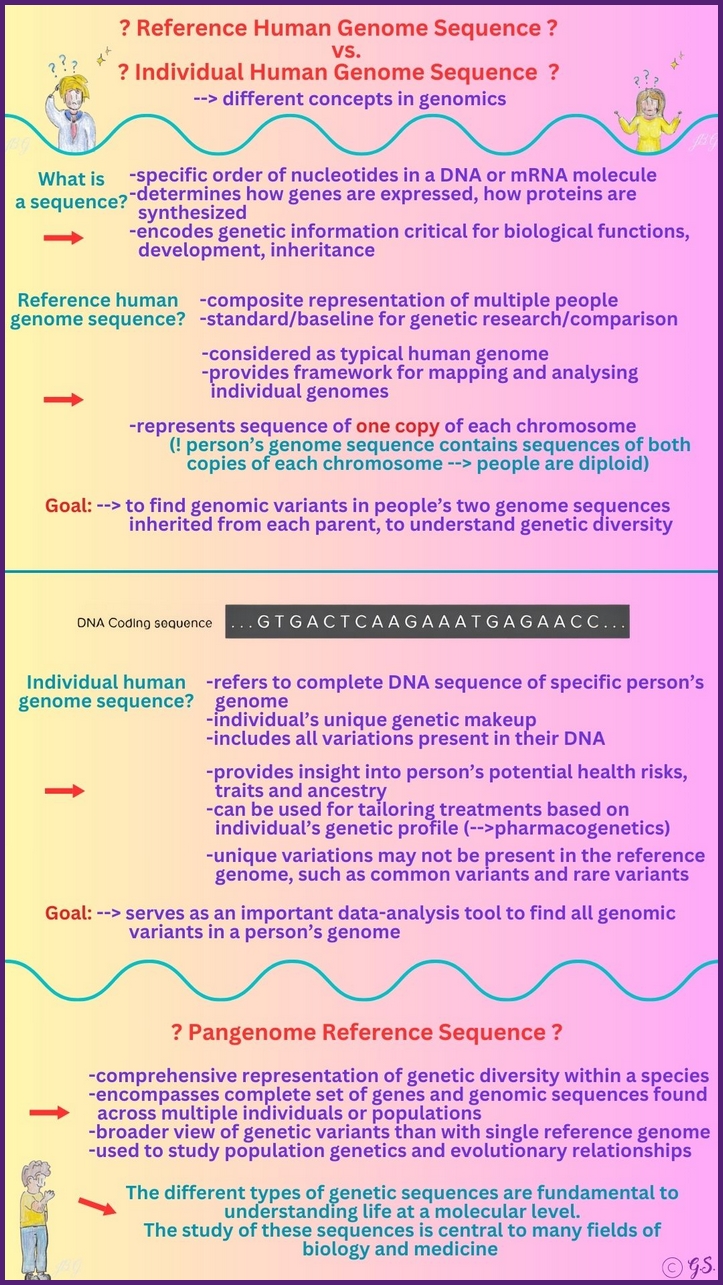
Was sind Biomaker?
Bislang gibt es keine Heilung für STXBP1-RD, aber gezielte Therapien für STXBP1 werden derzeit untersucht. Die Variabilität der Symptome und der Schweregrad von STXBP1-RD erschweren jedoch die Bewertung der Wirkung einer Behandlung im Rahmen klinischer Studien, sodass es sehr wichtig ist, messbare Biomarker für die Krankheit zu identifizieren.
Biomarker sind für STXBP1-Klinische Studien wichtig, weil sie helfen können, das Verständnis der Erkrankung zu verbessern, den Krankheitsverlauf zu überwachen und den Behandlungserfolg zu messen. STXBP1-Mutationen sind mit bestimmten schweren Epilepsieformen und neuroentwicklungsbezogenen Störungen assoziiert. Biomarker können frühere Prädiktoren für Ansprechen auf Therapien, Risikostufen für schwere Anfälle oder Entwicklungsverlauf darstellen. Sie ermöglichen "stratified medicine", also die Einteilung von Patienten in Untergruppen, die unterschiedlich auf Therapien reagieren könnten, und unterstützen so gezieltere, personalisierte Behandlungsansätze.
Zurzeit werden vor allem Biomarker aus Blut, Liquor, Hirnbildgebung oder neurologischen Tests identifiziert, standardisierte Assays entwickelt, Probenahme- und Analyseprotokolle harmonisiert und frühe Studien durchgeführt, die Biomarker mit Endpunkten wie Anfallhäufigkeit und Entwicklungsfortschritt verknüpfen. Internationale Konsortien bündeln Patientenkohorten, um die Aussagekraft zu erhöhen, und präklinische Modelle helfen bei der Validierung potenzieller Biomarker, bevor sie in Studien genutzt werden. Ziel ist die Etablierung zuverlässiger Biomarker als Einschlusskriterien, Surrogatmarker oder Sicherheitsindikatoren.

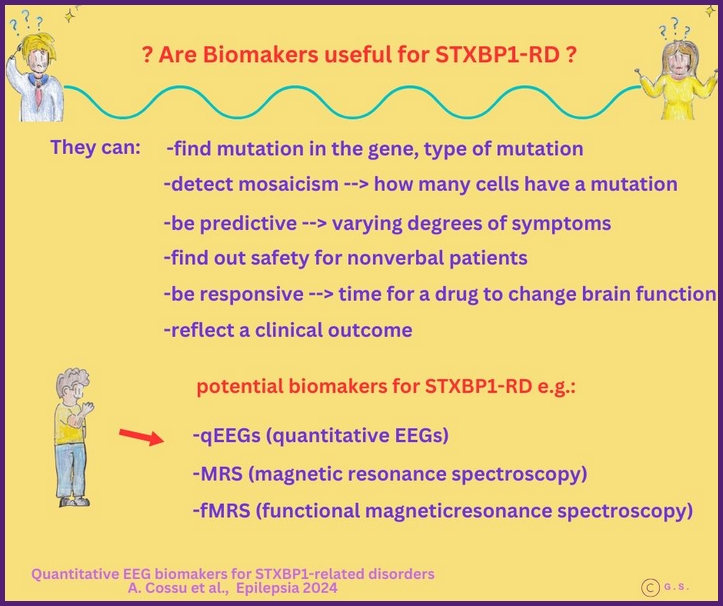
Gen-Therapie vs. Gen-Editierung vs. ASOs
Gen-Therapie und Gen-Editierung sind verwandte, aber unterschiedliche Ansätze zur Veränderung genetischer Informationen zur Behandlung oder Erforschung von Krankheiten.
Die Gen-Therapie zielt darauf ab, Krankheiten zu behandeln oder zu verhindern, indem genetisches Material in die Zellen eines Patienten eingebracht, hinzugefügt oder reaktiviert wird. Dabei werden häufig Vektoren wie virale Transportsysteme verwendet, um eine korrekte Kopie eines fehlerhaften Gens einzuführen oder therapeutische RNA oder genregulierende Elemente zu transportieren, mit dem Ziel, eine langfristige, systemische Veränderung der Genexpression zu erreichen.
Die Gen-Editierung hingegen konzentriert sich darauf, präzise, gezielte Veränderungen am bestehenden Genom vorzunehmen. Techniken wie CRISPR-Cas-Systeme ermäglichen es Forschern, DNA an bestimmten Stellen zu schneiden, wodurch Korrekturen, Deletionen oder Insertionen innerhalb des endogenen Gens möglich werden und somit das Genom selbst verändert wird.
Kurz gesagt, die Gen-Therapie neigt dazu, genetisches Material hinzuzufügen oder zu modifizieren, um eine therapeutische Wirkung zu erzielen, während die Gen-Editierung das Genom direkt verändert, um genetische Informationen zu korrigieren oder neu zu konfigurieren. Beide Strategien sind vielversprechend und bringen wichtige technische, ethische und sicherheitsrelevante Überlegungen mit sich, darunter die Effizienz der Verabreichung, Off-Target-Effekte, langfristige Haltbarkeit und behördliche Aufsicht.
ASOs (Antisense-Oligonukleotide) sind kurze, synthetische Nukleinsäurestränge, die spezifische RNA-Transkripte binden sollen. Durch die Paarung mit der Ziel-RNA können sie die Genexpression modulieren, ohne die DNA zu verändern.
ASOs werden eingesetzt, um die Produktion krankheitsverursachender Proteine zu reduzieren oder zu modifizieren, und finden Anwendung bei einer Reihe von genetischen Erkrankungen. Sie erfordern eine sorgfältige Verabreichung, chemische Modifikationen für Stabilität und Verträglichkeit sowie eine gründliche Bewertung von Off-Target-Effekten und der langfristigen Sicherheit.


Mitochondrien – mtDNA
Mitochondrien sind Organellen innerhalb von Zellen, die den größten Teil des ATP, der primären Energiequelle der Zelle, erzeugen. Sie enthalten ihre eigene ringförmige DNA und eine Doppelmembran und sind neben der Energieproduktion an zahlreichen weiteren wichtigen Prozessen beteiligt, darunter die Regulierung der Apoptose (programmierter Zelltod), die Speicherung von Kalzium und die kontrollierte Produktion reaktiver Sauerstoffspezies. Die meisten mitochondrialen Proteine werden von der Kern-DNA kodiert und in die Mitochondrien importiert, während ein Teil davon vom mitochondrialen Genom (mtDNA) kodiert wird.
Mutationen in den Mitochondrien können eine Vielzahl von Gesundheitsproblemen verursachen. Sie können die mitochondriale Atmungskette und die oxidative Phosphorylierung beeinträchtigen und so die ATP-Produktion verringern. Dieser Energiemangel wirkt sich auf viele Organe und Systeme aus. Häufige Symptome sind Muskelschwäche, Müdigkeit, Hörverlust, Sehstörungen, Herz-Kreislauf-Probleme und Nierenschäden. Neurologische Beeinträchtigungen können Epilepsie, Entwicklungsverzögerungen, Koordinationsstörungen und Neurodegeneration umfassen. Zu den metabolischen Manifestationen kann eine Laktatazidose gehören.
Im Zusammenhang mit STXBP1-RD (Munc18-1-bedingte neurologische Entwicklungsstörung) sind mitochondriale Aspekte relevant, da der Energiebedarf der Nervenzellen hoch ist und die mitochondriale Funktion die Widerstandsfähigkeit der Nervenzellen und die Anfallsanfälligkeit beeinflussen kann. Wenn Anzeichen für eine mitochondriale Beteiligung auftreten – wie anhaltende Laktatazidose, Hypoglykämie oder multisystemische Merkmale – können ein Stoffwechselprofil und gezielte mitochondriale Tests als Teil der Untersuchung in Betracht gezogen werden.
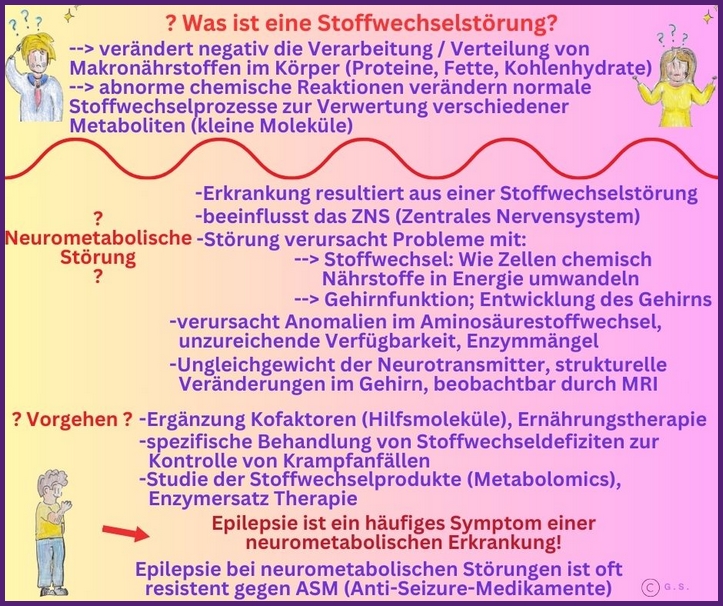
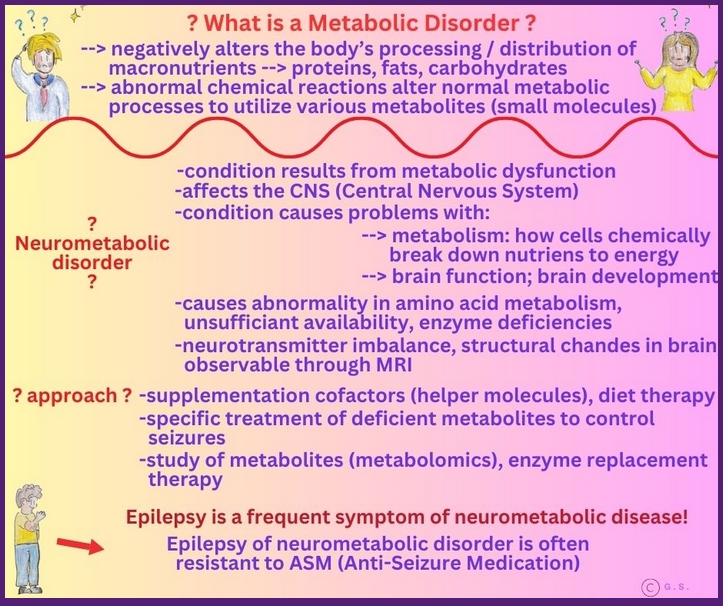
Diagnose vs. Syndrom
Eine klare Unterscheidung zwischen einer Diagnose und einem Syndrom ist wichtig, um zu verstehen, wie medizinische Informationen zur Beurteilung der Gesundheit und zur Steuerung der Behandlung verwendet werden.
Eine Diagnose benennt eine bestimmte Krankheit oder Störung mit einer bestimmten zugrunde liegenden Ursache oder Pathologie, die in der Regel durch die Erfüllung definierter Kriterien festgestellt und häufig durch Tests wie Laborergebnisse, Bildgebung oder genetische Analysen bestätigt wird. Sie zielt darauf ab, das Grundproblem oder den Grundzustand zu identifizieren, der die Symptome des Patienten erklärt.
Ein Syndrom hingegen beschreibt ein erkennbares Muster von Symptomen und Befunden, die tendenziell zusammen auftreten, aber mehrere mögliche Ursachen haben können. Es betont eher die Häufung oder das Erscheinungsbild als die Ermittlung einer einzigen Ursache. Beispiele hierfür sind das Down-Syndrom, das eine Reihe charakteristischer Merkmale bezeichnet, oder das metabolische Syndrom, das mehrere miteinander verbundene Stoffwechselstörungen zusammenfasst.
Kurz gesagt, eine Diagnose identifiziert eine bestimmte Krankheit oder Störung mit einer definierten Ursache, während ein Syndrom eine konsistente Sammlung von Symptomen beschreibt, die aus verschiedenen Grunderkrankungen entstehen können. Klinisch hilft diese Unterscheidung dabei, zu entscheiden, welche Tests angeordnet werden sollen, wie die Behandlung angepasst werden kann und wie die Prognose mit Patienten und Familien besprochen werden soll.


Forschungstechniken?
Forschungstechniken umfassen eine Reihe von Ansätzen, die darauf ausgelegt sind, Hypothesen in kontrollierten und realen Kontexten zu beobachten, zu messen und zu testen. Die wesentlichen Unterschiede ergeben sich daraus, wie Daten erzeugt und kontrolliert werden.
Experimentelle Methoden manipulieren aktiv Variablen, um Ursache-Wirkungs-Zusammenhänge herzustellen, während Beobachtungsmethoden natürliche Phänomene aufzeichnen, um Zusammenhänge zu identifizieren und Hypothesen zu generieren.
Im Rahmen der experimentellen Techniken werden bei In-vitro-Methoden Experimente außerhalb eines lebenden Organismus durchgeführt, häufig in Zellen, Geweben oder biochemischen Systemen, wodurch eine präzise Kontrolle der Umgebung und eine schnelle Prüfung von Mechanismen, Arzneimittelwirkungen oder zellulären Reaktionen ermöglicht wird. In-vitro-Ansätze werden für Vorab-Screenings, mechanistische Studien, Toxizitätstests und Optimierungen verwendet, bevor komplexere Modelle zum Einsatz kommen.
In-vivo-Techniken umfassen die Untersuchung von Prozessen innerhalb lebender Organismen und liefern Einblicke in die integrierte Physiologie, Pharmakokinetik und Ganzkörperreaktionen, die in isolierten Systemen nicht zugänglich sind. Tiermodelle, klinische Studien und Human-Organ-on-a-Chip-Systeme sind Beispiele für In-vivo- und Hybridansätze, die Laborergebnisse mit der realen Biologie verbinden.
In-silico-Ansätze verwenden Computersimulationen und Rechenmodelle, um Systeme zu untersuchen, wenn Experimente nicht praktikabel sind, oder um Laborarbeiten zu ergänzen.
Sie werden verwendet, um biologische Systeme, Medikamente oder Krankheiten ohne tatsächliche Laborexperimente oder lebende Organismen zu untersuchen. In-silico-Tests können die Forschung beschleunigen, Kosten senken und bei der Priorisierung von Experimenten helfen, aber die Ergebnisse müssen in der Regel experimentell validiert werden.
Bei allen Methoden hängt die Wahl der Technik von der Forschungsfrage, der Ebene der biologischen Organisation, ethischen Überlegungen und praktischen Faktoren wie Kosten, Zeit und Durchführbarkeit ab.

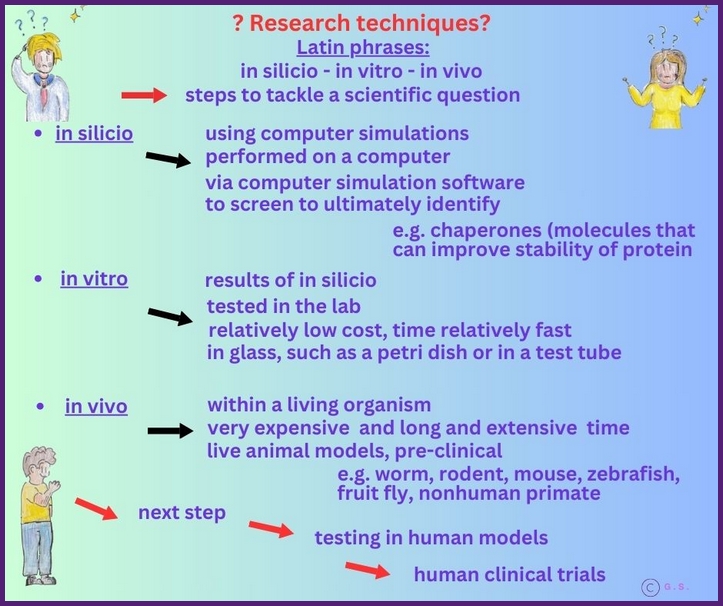
Was sind Chaperone?
Chaperone sind eine vielfätige Gruppe von zellulären Proteinen, die bei der korrekten Faltung, Stabilisierung und dem Transport anderer Proteine helfen. Sie unterstützen neu gebildete Polypeptide dabei, sich zu ihrer korrekten dreidimensionalen Struktur zu falten, verhindern Aggregationen und helfen bei der Rückfaltung oder dem Abbau fehlgefalteter Proteine.
Proteine können bei STXBP1 Fehlfunktionen aufweisen. Chaperone unterstützen die korrekte Faltung und Stabilisierung und eröffnen damit neue Ansatzpunkte für bereits zugelassene Medikamente. Als zelluläre Helfer leiten Chaperone Proteine dazu an, sich dann korrekt zu falten und an ihrem richtigen Platz zu bleiben. Durch die Stabilisierung fehlgefalteter oder instabiler Proteine eröffnen sie Möglichkeiten für die Umwidmung bestehender Medikamente zur Behandlung von Krankheiten, Drug Repurposing. In der gesamten Zelle überwachen Chaperone die Qualitätskontrolle von Proteinen, modulieren deren Stabilität und Interaktionen und verwandeln so bekannte Medikamente in Therapien für verschiedene Erkrankungen.
Im Zusammenhang mit STXBP1-Mutationen werden Chaperon-basierte Ansätze untersucht, um die Stabilität und Funktion des STXBP1-Proteins zu verbessern, das durch Missense-Mutationen destabilisiert oder fehlgefaltet werden kann. Durch die Bindung an das mutierte Protein können Chaperone die korrekte Faltung fördern, Aggregationen verhindern und dessen Reifung und Lokalisierung in Neuronen unterstützen, wodurch möglicherweise die Freisetzung synaptischer Vesikel und die neuronale Kommunikation wiederhergestellt werden.
Zu den Vorteilen dieser Strategie gehören das Potenzial, die Restaktivität bei Funktionsverlustmutationen zu retten, das therapeutische Fenster durch Stabilisierung instabiler Varianten zu erweitern und einen mutationsspezifischen Ansatz anzubieten, der den Genersatz oder die Genbearbeitung ergänzen oder ersetzen kann. Die Chaperon-Therapie kann auch die Belastung durch fehlgefaltete Proteine verringern und dadurch den zellulären Stress reduzieren und die Gesundheit der Neuronen verbessern.
Es gibt jedoch auch erhebliche Risiken und Herausforderungen. Chaperone können Off-Target-Effekte haben und nicht-funktionelle oder pathogene Konformationen in anderen Proteinen stabilisieren, was zu unbeabsichtigten zellulären Folgen führen kann. Es ist entscheidend, das richtige Maß an Stabilisierung zu erreichen; eine Überstabilisierung kann Proteine in unproduktiven Zuständen festhalten oder den normalen Umsatz stören.
Darüber hinaus kann die Wirksamkeit der Chaperon-Therapie in hohem Maße mutationsspezifisch sein, was ein genaues Verständnis der Faltungsdefekte jeder STXBP1-Variante erfordert. Eine sorgfältige präklinische Bewertung und ein biomarkerbasiertes Design klinischer Studien sind unerlässlich, um potenzielle Vorteile und Risiken gegeneinander abzuwägen.
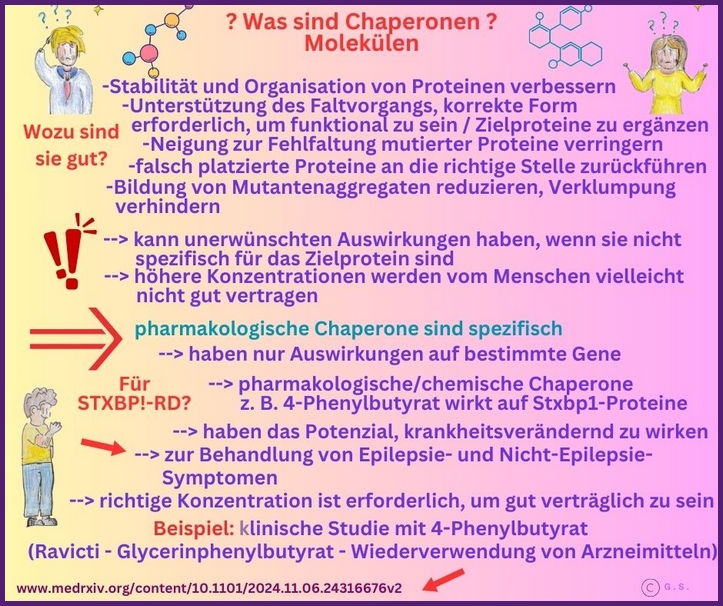
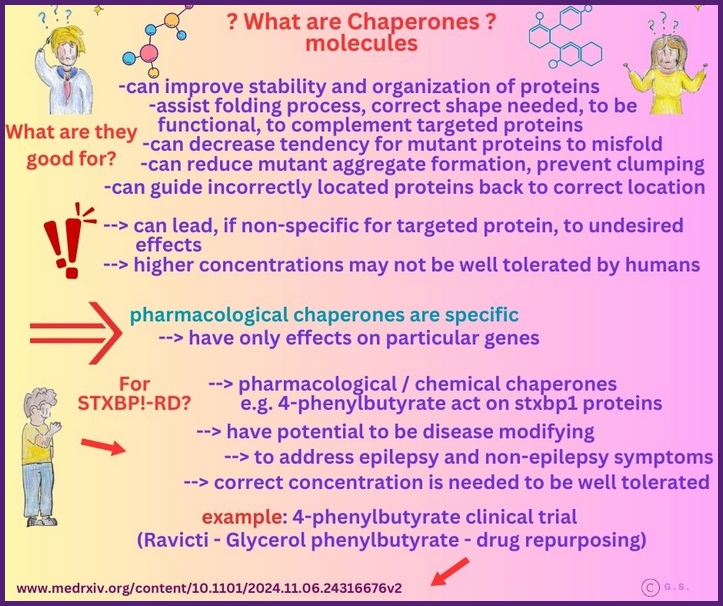
Genetik vs. Epigenetik?
Genetik und Epigenetik sind zwei eng miteinander verbundene Informationsebenen, die bestimmen, wie Organismen sich entwickeln, funktionieren und auf ihre Umgebung reagieren. Die Genetik befasst sich mit den Genen selbst – ihren Sequenzen, Varianten und der Frage, wie Veränderungen in der DNA vererbte Merkmale kodieren. Sie erklärt, warum Menschen bestimmte Eigenschaften von ihren Eltern erben und wie Mutationen zu einer Veranlagung für Krankheiten führen können.
Die Epigenetik hingegen untersucht, wie die Genaktivität reguliert wird, ohne die zugrunde liegende DNA-Sequenz zu verändern. Zu den epigenetischen Mechanismen gehören chemische Modifikationen der DNA und der Histonproteine, wie Methylierung und Acetylierung, sowie die Wirkung nicht-kodierender RNAs. Diese Markierungen können Gene aktivieren oder deaktivieren, die Zellidentität prägen und auf Umwelteinflüsse wie Ernährung, Stress und Toxine reagieren. Wichtig ist, dass epigenetische Veränderungen dynamisch und in einigen Fällen reversibel sein können, wodurch sie die Lücke zwischen dem Genom eines Organismus und seiner sich entwickelnden Physiologie und seinem Verhalten schließen.
Sowohl die Genetik als auch die Epigenetik sind für das Verständnis von Gesundheit und Krankheit von entscheidender Bedeutung: Die Genetik liefert den Bauplan, während die Epigenetik erklärt, wie dieser Bauplan in verschiedenen Kontexten gelesen und umgesetzt wird. Für Forscher bietet die Erforschung der Epigenetik Einblicke in Entwicklungsprozesse, Alterung und Tumorentstehung und kann Ansatzpunkte für Interventionen aufzeigen, selbst wenn die DNA-Sequenz unverändert bleibt.


Was ist der Unterschied zwischen RWD und RWE?
Bedeutung von Real-World-Daten (RWD) und Real-World-Evidence (RWE)
RWD sind gesundheitsbezogene Daten, die aus Quellen außerhalb traditioneller randomisierter kontrollierter Studien stammen, wie z. B. elektronische Gesundheitsakten, Patientenregister, Abrechnungsdaten und von Patienten berichtete Ergebnisse, während RWE das umsetzbare Wissen ist, das aus der Analyse dieser Daten gewonnen wird, um zu verstehen, wie Behandlungen in der Praxis wirken.
Der Unterschied liegt in den Daten gegenüber den Erkenntnissen: RWD sind das Rohmaterial, und RWE sind die Schlussfolgerungen und Interpretationen, die aus der Analyse dieses Materials gezogen werden.
Beide sind wichtig, da RWD und RWE randomisierte Studien ergänzen, indem sie verschiedene Patientengruppen, längere Zeithorizonte und reale Anwendungsmuster widerspiegeln, was dabei hilft, die Wirksamkeit, Sicherheitssignale und den Wert in der Routineversorgung zu ermitteln. Zusammen liefern sie Informationen für die Entscheidungsfindung von Klinikern, Forschern, politischen Entscheidungsträgern und Patienten und dienen als Leitfaden für Behandlungsentscheidungen, die Überwachung nach der Markteinführung, die Bewertung von Gesundheitstechnologien und die Zuweisung von Ressourcen.


Warum STXBP1 Klinische Studien?
Klinische Studien sind systematische Untersuchungen, in denen getestet wird, ob neue Behandlungen, Geräte oder Strategien bei Menschen sicher und wirksam sind. Sie folgen einem vordefinierten Plan, um bestimmte Fragen zu beantworten, beispielsweise wie gut eine Therapie wirkt, welche Nebenwirkungen sie verursachen kann und wie sie im Vergleich zur Standardbehandlung abschneidet. Studien durchlaufen verschiedene Phasen, die jeweils einen unterschiedlichen Zweck verfolgen und unterschiedliche Nachweise erfordern, um fortgesetzt werden zu können.
In Phase-I-Studien werden die Sicherheit und Dosierung an einer kleinen Gruppe von Teilnehmern getestet, wobei der Schwerpunkt darauf liegt, wie der Köper auf die Behandlung reagiert und welche Nebenwirkungen auftreten können.
In Phase-II-Studien wird die vorläufige Wirksamkeit untersucht und die Sicherheit in einer größeren Gruppe weiter bewertet, um die optimale Dosis oder Dosierung zu ermitteln.
In Phase-III-Studien wird die neue Behandlung mit der aktuellen Standardbehandlung in einer großen, vielfältigen Population verglichen, um die Gesamtwirksamkeit zu bestimmen, seltene Nebenwirkungen zu überwachen und Informationen zu sammeln, die die behördliche Zulassung unterstützen.
Phase-IV-Studien finden nach der Zulassung und Markteinführung einer Behandlung statt. Sie überwachen die langfristige Sicherheit, die Wirksamkeit in der Praxis und die Auswirkungen auf die Ergebnisse einer größeren Patientengruppe.
Die Zulassung durch Aufsichtsbehörden wie die FDA (US-amerikanische Lebensmittel- und Arzneimittelbehörde) oder die EMA (Europäische Arzneimittelagentur) erfolgt, nachdem erfolgreiche Phase-III-Studien (und manchmal auch Phase-II-Daten) gezeigt haben, dass der Nutzen die Risiken überwiegt. Die Behörden prüfen das gesamte Datenpaket, einschließlich Sicherheit, Wirksamkeit, Herstellungsqualität und Kennzeichnung, bevor sie die Marktzulassung erteilen. Die Überwachung nach der Zulassung (Phase IV) dient weiterhin der Überwachung der Sicherheit und Wirksamkeit in der Praxis.
Dieser Verlauf – von der anfänglichen Sicherheit bis zur umfassenderen Bestätigung des Nutzens – ermöglicht es den Forschern, solide Beweise zu sammeln und gleichzeitig die Teilnehmer zu schützen.


STXBP1 Kinder haben Schlafprobleme
Schlaf ist für das Gehirn von entscheidender Bedeutung, auch bei Kindern mit Erkrankungen wie STXBP1-bedingten Störungen. Während des Schlafs festigt das Gehirn Erinnerungen und Lernerfahrungen des Tages. Tiefschlaf (Non-REM) unterstützt die körperliche und kognitive Erholung, während REM-Schlaf die emotionale Verarbeitung und kreative Problemlösung fördert. Schlaf unterstützt auch Reinigungsprozesse im Gehirn, die Abfallprodukte beseitigen und so die langfristige Gesundheit der Nervenzellen schützen.
Schlafprobleme können die Aufmerksamkeit, das Verhalten, das Lernen und das Ansprechen auf Therapien beeinträchtigen. Die Kontrolle von Anfällen, die Regulierung der Stimmung und die Reaktion auf Medikamente können durch schlechten Schlaf beeinträchtigt werden.


Natural History Studies
Natural History Studies zur STXBP1-Erkrankung sind äußerstwichtig, da sie dokumentieren, wie sich die Erkrankung ohne experimentelle Intervention im Laufe der Zeit typischerweise entwickelt. Sie liefern eine Grundlage für das Verständnis typischer Verläufe der neurologischen Entwicklung, der Anfallshäufigkeit, motorischer und sprachlicher Meilensteine, Verhaltensmerkmale und Komorbiditäten wie geistige Behinderung, Merkmale des Autismus-Spektrums und Schlafstörungen. Durch die Erfassung der Variabilität zwischen einzelnen Personen und verschiedenen Altersgruppen hilft die Naturgeschichtsforschung dabei, kritische Zeitfenster für die Behandlung, Prognose und Lebensqualität zu identifizieren. Durch die systematische Beobachtung von Symptomen, Verlauf und Reaktionen auf Standardbehandlungen in der Praxis zeigen diese Studien Muster auf, die randomisierte Studien allein nicht erfassen können, wie z. B. langfristige Komplikationen, natürliche Remissionen und Faktoren, die den Krankheitsverlauf beeinflussen. Sie unterstützen auch die Entwicklung und Validierung aussagekräftiger klinischer Endpunkte. Die Naturgeschichtsforschung ist oft der erste Schritt zur Erkennung von Subtypen.
Bei der STXBP1-Erkrankung, bei der die Phänotypen breit gefächert sind und sie sich im Laufe der Zeit weiterentwickeln können, sind Naturgeschichtsdaten besonders wertvoll, um Genotyp-Phänotyp-Muster zu erkennen, Diagnosekriterien zu verfeinern und Forschungsfragen zu priorisieren, die den Patienten am meisten zugutekommen. Letztendlich bildet diese Forschung die Grundlage für eine evidenzbasierte Versorgung und gemeinsame Anstrengungen zur Verbesserung der Überwachung, Beratung und Therapien während des gesamten Lebens von Menschen mit STXBP1-Erkrankung. Diese Studien sind für klinische Studien wichtig, da sie die grundlegenden Krankheitsverläufe definieren, natürliche Variabilität identifizieren, aussagekräftige Endpunkte und Ergebnismaße festlegen, bei der Berechnung der Aussagekraft und der Schätzung der Stichprobengröße helfen und eine Referenz liefern, anhand derer die Wirkungen experimenteller Therapien beurteilt werden können. Sie liefern auch Informationen für die Patientenauswahl, die Stratifizierung nach Subtypen und die Interpretation von Sicherheitsdaten in Studien und stellen so sicher, dass die Ergebnisse auf die vielfätige STXBP1-Population anwendbar sind.


Gentests für alle!
Weltweit gibt es so viele Kinder, die aufgrund des begrenzten Zugangs zu Gentests und mangelnder Aufklärung nicht diagnostiziert sind.
Eine frühzeitige Diagnose durch Gentests ist für eine angemessene Behandlung und Unterstützung von entscheidender Bedeutung.
Eine verstärkte Aufklärung und Ausweitung der Testmaßnahmen kann dazu beitragen, diese Kinder früher zu identifizieren und ihnen eine bessere Versorgung und bessere Behandlungsmöglichkeiten zu bieten.
EEG gibt es in verschiedenen Formen
Die Elektroenzephalographie (EEG) ist ein wichtiges Instrument in der Neurologie zur Aufzeichnung der elektrischen Aktivität des Gehirns und für die Beurteilung von Epilepsie unverzichtbar.
Im Laufe der Jahre haben sich die EEG–Techniken von kurzen Standardaufzeichnungen zu längeren, multimodalen Ansätzen entwickelt, darunter erweiterte Überwachung und Videointegration sowie spezielle Methoden während des Schlafs oder unter provokativen Bedingungen. Diese vielfältigen Formen helfen Ärzten dabei, epileptiforme Aktivitäten zu erkennen, Anfälle zu klassifizieren, epileptogene Zonen zu lokalisieren und das Ansprechen auf Behandlungen zu überwachen.
Obwohl das EEG von unschätzbarem Wert ist, ist es nur ein Teil des diagnostischen Puzzles und sollte im Zusammenhang mit der Krankengeschichte und anderen Untersuchungen interpretiert werden.
Heute wird das EEG mit Bildgebung, Neuropsychologie und Genetik kombiniert, und automatisierte Analysen sowie maschinelles Lernen unterstützen zunehmend die Interpretation.


|