STXBP1 Forschungsansätze
STXBP1 Forschungsansätze |
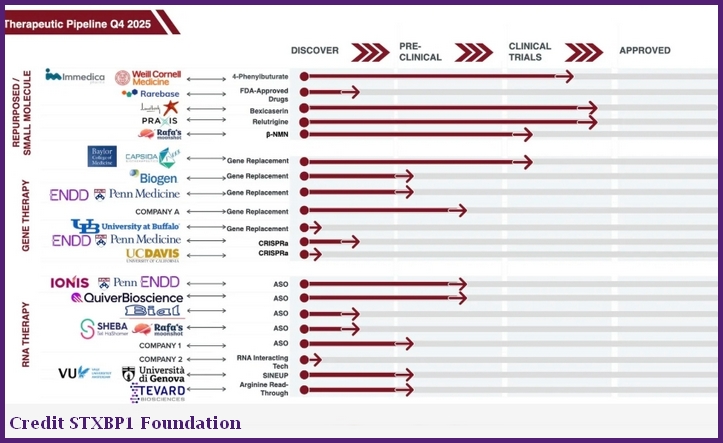
STXBP1 Pipelines Repurposed drugs, Gentherapien und RNA Therpien |
Was sind Chaperonen?
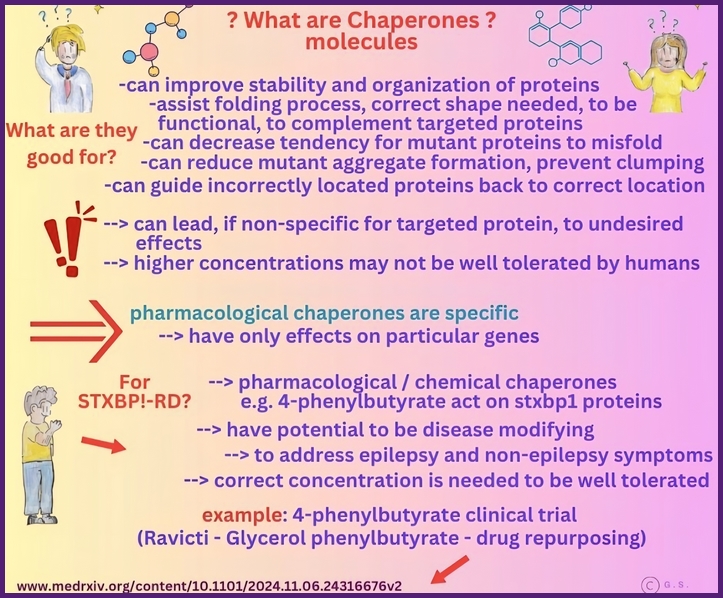
Chaperone sind eine vielfältige Gruppe von zellulären Proteinen, die bei der korrekten Faltung, Stabilisierung und dem Transport anderer Proteine helfen. Sie unterstützen neu gebildete Polypeptide dabei, sich zu ihrer korrekten dreidimensionalen Struktur zu falten, verhindern Aggregationen und helfen bei der Rückfaltung oder dem Abbau fehlgefalteter Proteine.
Im Zusammenhang mit STXBP1-Mutationen werden Chaperon-basierte Ansätze untersucht, um die Stabilität und Funktion des STXBP1-Proteins zu verbessern, das durch Missense-Mutationen destabilisiert oder fehlgefaltet werden kann. Durch die Bindung an das mutierte Protein können Chaperone die korrekte Faltung fördern, Aggregationen verhindern und dessen Reifung und Lokalisierung in Neuronen unterstützen, wodurch möglicherweise die Freisetzung synaptischer Vesikel und die neuronale Kommunikation wiederhergestellt werden.
Zu den Vorteilen dieser Strategie gehören das Potenzial, die Restaktivität bei Funktionsverlustmutationen zu retten, das therapeutische Fenster durch Stabilisierung instabiler Varianten zu erweitern und einen mutationsspezifischen Ansatz anzubieten, der den Genersatz oder die Genbearbeitung ergänzen oder ersetzen kann. Die Chaperon-Therapie kann auch die Belastung durch fehlgefaltete Proteine verringern und dadurch den zellulären Stress reduzieren und die Gesundheit der Neuronen verbessern.
Es gibt jedoch auch erhebliche Risiken und Herausforderungen. Chaperone können Off-Target-Effekte haben und nicht-funktionelle oder pathogene Konformationen in anderen Proteinen stabilisieren, was zu unbeabsichtigten zellulären Folgen führen kann. Es ist entscheidend, das richtige Maß an Stabilisierung zu erreichen; eine Überstabilisierung kann Proteine in unproduktiven Zuständen festhalten oder den normalen Umsatz stören.
Darüber hinaus kann die Wirksamkeit der Chaperon-Therapie in hohem Maße mutationsspezifisch sein, was ein genaues Verständnis der Faltungsdefekte jeder STXBP1-Variante erfordert. Eine sorgfältige präklinische Bewertung und ein biomarkerbasiertes Design klinischer Studien sind unerlässlich, um potenzielle Vorteile und Risiken gegeneinander abzuwägen.
Funktion von STXBP1 in nicht-neuronalen Zellen?
Die STXBP1 Foundation hat Helen Willsey, PhD, von der University of California, San Francisco, einen Zuschuss von bis zu 100.000 US-Dollar gewährt. In ihrer Forschung untersucht sie anhand von Froschmodellen neurologische Störungen.
Sie erforscht die Funktionen von STXBP1 außerhalb des Gehirns, insbesondere in Flimmerzellen.
Bislang wurden nur aktive Neuronen oder nur Gehirnzellen untersucht. Diese Studie geht davon aus, dass STXBP1 auch in anderen Zellen als denen des Gehirns aktiv ist, insbesondere in Flimmerzellen.
Durch die Untersuchung, wie STXBP1 zur Gesundheit der Atemwege beiträgt, hofft sie, neue Erkenntnisse darüber zu gewinnen, warum Atemkomplikationen nach wie vor eine der Hauptursachen für vorzeitige Sterblichkeit bei Menschen mit STXBP1-bedingten Erkrankungen sind.
Atemwegszilien oder Flimmerzellen sind spezialisierte Zellen, die haarähnliche Strukturen besitzen.
Sie befinden sich in den Atemwegen, die eine entscheidende Rolle im Abwehrmechanismus des Körpers spielen. Sie sind Teil des pseudostratifizierten zylindrischen Flimmerepithels, das die Atemwege von der Nasenhöhle bis zu den Bronchiolen auskleidet.
Diese Studie könnte neue Erkenntnisse über die Funktion von STXBP1 in nicht-neuronalen Zellen liefern.
Dr Willsey's work
STXBP1 Foundation Awards Innovation Grant to Helen Willsey for Gene Therapy Development
L-Serin als STXBP1-Therapie?
L-Serin ist eine nicht-essentielle Aminosäure, die im Körper gebildet wird und auch über die Nahrung aufgenommen werden kann. Sie spielt eine Rolle als Vorstufe in verschiedenen Stoffwechselwegen, einschließlich der Synthese von Proteinen, Neurotransmittern und Membranlipiden.
L-Serin ist auch Gegenstand medizinischer Forschung, insbesondere im Zusammenhang mit neurologischen Syndromen. Eine L-Serin-Therapie ist eine medizinische Behandlung, bei der die Aminosäure L-Serin – allgemein als sicher eingestuft – meist in Form von Nahrungsergänzungsmitteln, zur Unterstützung eingesetzt wird.
L-Serin ist wichtig für die Produktion von Neurotransmittern, den Aufbau von Nervengewebe, die kognitive Leistungsfähigkeit und fördert die Muskelgesundheit.
L-Serin-Therapien werden auch erforscht zur Behandlung von neurodegenerativen Erkrankungen. L-Serin ist entscheidend für die Bildung von Phosphatiden und Sphingosin, wichtige Bestandteile von Nerven- und Gehirnzellen. Er unterstützt die Produktion von Neurotransmittern wie Acetylcholin und ist wichtig für die Signalübertragung im Gehirn.
L-Serin ist derzeit noch kein zugelassenes Medikament. Es wird deshalb abgeraten, L-Serin eigenmächtig zur Behandlung von neurodegenerativen Erkrankungen einzunehmen, bis weitere Forschungsergebnisse vorliegen.
Krankheitsbilder, die mit LOF-Varianten – wie oft bei STXBP1 – potenziell einer Therapie mit L-Serin zugänglich sind, werden diskutiert. L-Serin ist ein Vorläufer für D-Serin und beteiligt an der Synthese von D-Serin, das als Coagonist des NMDA-Rezeptors wirken kann. Die Modulation des NMDA-Tonus könnte die synaptische Transmission beeinflussen, die neuronale Funktion unterstützen, die metabolische Blockaden lindern und Auswirkungen auf synaptische Plastizität und Epilepsie haben.
Als Baustein fü,r Lipide und Proteine kann L-Serin über verschiedene Wege die Membran- und Synapsenbiologie beeinflussen, was bei Störungen der Vesikel-Fusion theoretisch unterstützend wirken könnte. Die existierende Evidenzbasis für L-Serin bei STXBP1 ist aber noch begrenzt und überwiegend theoretisch oder präklinisch (Tiermodelle, Zellmodelle) bzw. allgemeiner Natur (Epilepsie-/Neurotransmissions-Kontext), ohne klare klinische Richtlinien. Wirkung, Sicherheit und Dosierung von L-Serin können individuell variieren; eine unsachgemäße Anwendung kann Risiken bergen.
Quelle: Genetik und genetische Diagnostik fokaler Epilepsien des Kindesalters, Dr. med. Ilona Krey et al., Clinical Epileptologie, Ausgabe 1/2024
Bexicaserin als STXBP1-Therapie?
Bexicaserin ist eine Substanz, ein Prüfpreparat, das als potenzieller Modulator im Nervensystem untersucht wird. Es wird vorrangig im Kontext von Neurotransmissionsregulation erforscht, doch der genaue zellulären Wirkmechanismus ist nicht eindeutig etabliert.
Bexicaserin könnte theoretisch Einfluss auf SNARE-Komplexe haben, wodurch das Gleichgewicht exzitatorische versus inhibitorische Signale verschoben werden könnte. Deshalb wird
Bexicaserin auch im Zusammenhang mit STXBP1-RD (DEEs) untersucht, aber es ist noch kein zugelassenes Medikament.
Es wurde von Longboard Pharmaceuticals entwickelt und von Lundbeck übernommen. Bexicaserin ist Teil der DEEpOCEAN-Studie, einer Phase-3-Studie für DEEs zu denen auch STXBP1-RD gehört. In einer PACIFIC-Studie (Phase 1b/2a) wurde Bexicaserin (auch: LP352) bei DEE-Patient:innen untersucht, mit positiven Ergebnissen für motorische Anfälle.
Das Medikament zeigt ein günstiges Sicherheits- und Verträglichkeitsprofil. Häufige Nebenwirkungen waren Infekte der oberen Atemwege, verringerter Appetit und Müdigkeit. Bexicaserin hat von der FDA eine Breakthrough Therapy Designation für DEEs erhalten. Es braucht weitere Studien (kontrollierte Phase-3- Studien), um Wirksamkeit, Sicherheit und Langzeitwirkung bei verschiedenen DEEs zu bestätigen.
Die DEEpOCEAN-Studie (Phase 3) schließt auch STXBP1-Patienten ein und wird global vorangetrieben, und startete auch in Deutschland, mit Startdatum: 08.10. 2025.
Bei Interesse an einer Studienteilnahme kann man sich bei den deutschen Studienzentren (Bielefeld, Kiel, Frankfurt, Ravensburg, Radeberg oder Bonn) melden. Kontakt:
DEEpOCEAN Studie
Clinical Trial DEEpOCEAN Info
Relutrigin als STXBP1-Therapie?
Relutrigin ist ein experimenteller Wirkstoff, ein erstklassisches kleines Molekül, das derzeit für die Behandlung bestimmter genetischen Epilepsieformen erforscht wird. Es wird auch eine mögliche Anwendung bei STXBP1-RD geprüft.
Relutriging wirkt als Präzisionsmodulator von Natriumkanälen. Natriumkanäle sind für die elektrische Aktivität von Nervenzellen wichtig. Relutrigin könnte dazu beitragen, die neuronale
Übereregbarkeit zu regulieren und somit Anfälle zu reduzieren. Die Anwendung von Relutrigin bei STXBP1-RD ist derzeit (Prax-562) noch ein Forschungsansatz und wird in klinischer Studie geprüft.
Relutrigin wurde in der EMBOLD-Studie zur Behandlung von Anfällen untersucht. Neben der Anfallskontrolle
wurde auch über Verbesserungen in anderen Bereichen wie Wachheit, Verhalten und Kommunikation berichtet. Die Embold-Studie der Praxis Precision Medicines Inc. ist eine multizentrische, doppelblinde,
placebokontrollierte, randomisierte Studie, die ein positives krankheitsmodifizierendes Potenzial in der Phase-2-Studie zeigte.
Die EMERALD-Studie ist eine Phase-3, randomisierte, placebokontrollierte Studie, die Wirksamkeit, Sicherheit, Verträglichkeit und Pharmakokinetik von Relutrigin, dasselbe Prüfpräparat wie in der EMBOLD-Studie, untersucht. Derzeit gibt es aktive Standorte in den Vereinigte Staaten, Südamerika, Europa, Vereinigtes Königreich, Australien. Es kann zwischen einer Teilnahme zu Hause, in der Klinik oder einer Kombination aus beiden gewählt werden. Praxis Precision Medicines Inc. sponsert die EMERALD-Studie. Infos:
EMERALD-Studie Clinical Trial
EMERALD-Studie Resilience Studies
EMERALD-Studie Press Release
ßNMN als STXBP1-Therapie?
"ßNMN Forschung" bezieht sich auf die wissenschaftliche Untersuchung von
Beta-Nicotinamid-Mononukleotid (abgekürzt als ß-NMN oder oft einfach nur NMN).
ß-NMN ist eine natürlich vorkommende Verbindung und eine Form von Vitamin B3 (Nicotinamid). Sie dient als direkte Vorstufe für Nicotinamid-Adenin-Dinukleotid (NAD+), ein essentielles Molekül, das in jeder Zelle des Körpers vorkommt und für grundlegende zelluläre Funktionen entscheidend ist.
Die Sicherheit von NMN als Nahrungsergänzungsmittel ist jedoch noch nicht nachgewiesen und die Substanz ist in vielen Ländern nur für Forschungszwecke zugelassen. In der EU einschließlich Deutschland gilt NMN als Novel Food und ist auch nicht als Nahrungsergänzungsmittel zugelassen.
Der Hauptfokus der Forschung liegt auf der Rolle von NMN bei Alterungsprozessen und altersbedingten Krankheiten.
Allerdings wurde in Tierstudien (Mäusen) auch eine Verbesserung des Energiestoffwechsels, die Reparatur von DNA-Schäden, den Schutz des Herzens und die Verlangsamung des kognitiven Verfalls festgestellt. ßNMN könnte die Gehirnfunktion durch die Unterstützung von DNA-Reparatur und die Reduzierung von Entzündungen fördern.
Klinische Studien am Menschen untersuchen die Sicherheit und Wirksamkeit von ßNMN zur Verbesserung der körperlichen Leistungsfähigkeit und anderer Gesundheitsparameter. Frühe Ergebnisse deuten auf eine gute Verträglichkeit und potenzielle Vorteile hin, aber es fehlen noch solide Langzeitstudien zur Sicherheit, Wirksamkeit und optimalen Dosierung.
In Israel wird Rafa001 (Beta-NMN) als Repuposing Drug untersucht. Bisher zeigte die Forschung positive Ergebnisse bei C. elegans, bei aus Patienten gewonnenen Neuronen und einem Mausmodell, was das Potenzial von ßNMN als schnell wirksames Therapeutikum untermauerte. Info:
Rafa's Moonshot ßNMN Pipeline
Was ist RNA Interacting Tech?
RNA Interacting Tech (Technologie zur Interaktion mit RNA) bezieht sich auf eine Reihe innovativer biochemischer und molekularbiologischer Methoden, die darauf abzielen, die Funktionen von Ribonukleinsäure (RNA) gezielt zu beeinflussen oder zu nutzen. Diese Technologien machen sich die zentrale Rolle der RNA im Körper zunutze, insbesondere als Botenmolekül (mRNA), das genetische Anweisungen von der DNA zur Proteinproduktion übermittelt.
Die bekanntesten Anwendungsformen sind RNA-Interferenz (RNAi) und Gen-Stilllegung. Dies ist eine der prominentesten RNA-interagierenden Technologien. Sie nutzt den natürlichen zellulären Mechanismus der Genregulation.
Kleine, synthetisch hergestellte RNA-Moleküle &lüar;wie small interfering RNAs, siRNAs) werden in die Zelle eingebracht. Dort binden sie an spezifische Ziel-mRNA-Moleküle und veranlassen deren Abbau oder blockieren deren Übersetzung in Proteine.
Diese Methode ermöglicht es, gezielt die Produktion von krankheitsverursachenden Proteinen zu stoppen, z.B. bei genetischen Krankheiten oder bestimmten Augenerkrankungen (Makuladegeneration).
Die Forschung konzentriert sich insbesondere auf die Entwicklung von Therapien, die auf RNA-Interferenz (RNAi) basieren, um Gene gezielt auszuschalten und Krankheiten zu behandeln. Info:
Fast Forward Pipelines
CRISPR-Cas9 vs. CRISPRa
CRISPR-Cas9 ist eine leistungsstarke Technologie zur Genbearbeitung, mit der Wissenschaftler präzise Veränderungen an der DNA vornehmen können. Sie besteht aus zwei Schlüsselkomponenten: einem Cas9-Enzym, das wie eine molekulare Schere wirkt, die die DNA schneidet, und einer Leit-RNA (gRNA), die Cas9 zu einer bestimmten DNA-Sequenz leitet. Wenn das Cas9-Enzym an der Zielstelle einen Doppelstrangbruch erzeugt, können die natürlichen Reparaturmechanismen der Zelle genutzt werden, um ein Gen zu deaktivieren, eine Mutation zu korrigieren oder ein neues Stück genetisches Material einzufügen. Das System wird wegen seiner Einfachheit, Flexibilität und relativ geringen Kosten im Vergleich zu früheren Genom-Editierungsmethoden geschätzt.
CRISPRa, oder CRISPR-Aktivierung, ist eine Variante des CRISPR-Systems, bei der ein katalytisch inaktives Cas9-Protein (dCas9) verwendet wird, das mit Transkriptionsaktivatoren fusioniert ist. Da das Cas9-Enzym bei der CRISPR-Cas9-Bearbeitung in der Lage ist, DNA zu schneiden, kann das dCas9 von CRISPRa keine DNA schneiden. Stattdessen bindet es sich unter der Anleitung der gRNA an die Promotorregion eines Gens oder andere regulatorische Elemente und rekrutiert den Transkriptionsapparat der Zelle, um die Expression dieses Gens zu erhöhen. Kurz gesagt, CRISPR-Cas9 editiert den genetischen Code durch Schneiden und Modifizieren der DNA, während CRISPRa die Genexpression moduliert, ohne die zugrunde liegende DNA-Sequenz zu verändern.
CRISPR-Cas9 wird für eine Vielzahl von Anwendungen eingesetzt, von der Erzeugung von Gen-Knockouts in Modellorganismen über die Korrektur krankheitsverursachender Mutationen in Zellen bis hin zum Screening auf Genfunktionen und der Entwicklung von Organismen für die Forschung oder therapeutische Entwicklung. CRISPRa wird verwendet, um Gene hochzuregulieren, um ihre Funktion zu untersuchen, um möglicherweise eine mangelhafte Genaktivität bei Krankheiten auszugleichen und um die Auswirkungen einer erhöhten Genexpression auf kontrollierte Weise zu untersuchen.
Zu den Vorteilen von CRISPR-Cas9 gehören seine relative Einfachheit, Genauigkeit und Vielseitigkeit, die gezielte Bearbeitungen in vielen Organismen und Zelltypen ermöglichen. Zu den potenziellen Risiken gehören Off-Target-Schnitte, bei denen unbeabsichtigte Teile des Genoms bearbeitet werden, unbeabsichtigte Immunreaktionen auf das bakterielle Cas9-Protein in klinischen Kontexten und ethische Bedenken hinsichtlich Keimbahnveränderungen, die vererbt werden könnten.
CRISPRa birgt weniger Risiken im Zusammenhang mit Störungen der DNA-Sequenz, da es das Genom nicht verändert, aber es kann durch Veränderung der Expressionsniveaus zu unbeabsichtigten Veränderungen in den Gen-Netzwerken führen, und die langfristigen Auswirkungen werden noch untersucht. Insgesamt sind beide Technologien für Forschungs- und Therapieanwendungen vielversprechend, aber ihre Anwendung erfordert eine sorgfältige Planung, gründliche Validierung und angemessene ethische und regulatorische Überlegungen.
Bei ENDD entwickelt eine Gruppe neue Werkzeuge, um die Expression der "gut" funktionierenden Kopie von STXBP1 mithilfe einer Anpassung der CRISPR-Cas9-Technologie namens CRISPR-Aktivierung (CRISPRa) hochzuregulieren. Info:
CRISPR-related Therapies
Center for Epilepsy and Neurodevelopmental Disorders
Gene Replacement Therapy – Genersatztherapie
Gene Replacement, auch als Genersatz bekannt, bezeichnet den Prozess, bei dem eine fehlfunktionierende oder fehlende Genlation durch eine funktionelle Kopie des Gens ersetzt wird. In der Forschung wird dies oft verwendet, um zu untersuchen, ob die Bereitstellung einer funktionellen STXBP1-Kopie Defekte in Synapsenfunktionen, Neurotransmitterfreisetzung oder epileptische Phänotypen bei STXBP1-Mutationen korrigieren kann. Typischerweise erfolgt der Ersatz durch Vektor-basierten Transfer, zum Beispiel mit Adeno-assoziierten Virusvektoren (AAV) oder anderen Vektoren, die eine korrekte Expression des STXBP1-Gens in Zielzellen ermöglichen. In Labormodellen, einschließlich Zellen aus iPSC-Derivaten oder transgenen Tiermodellen, wird geprüft, ob die eingeführte funktionelle Version des Gens Proteinproduktion und Funktionspfade wie Vesikelfusion und Synapseninaktivität wiederherstellt.
Es gibt Beispiele in der STXBP1-Forschung, in denen Forscher STXBP1-Expression durch virale Vektoren erhöht haben, um zu testen, ob eine erhöhte STXBP1-Expression Defizite mildern kann. Solche Ansätze dienen oft als präklinische Modelle zur Bewertung der Machbarkeit einer Genersatztherapie, bevor man zu klinischen Studien übergeht. Wichtig sind dabei die Wahl des richtigen Linups von Promotoren, die auf neuronale Zielzellen fokussiert sind, die Kontrolle der Expression, um Überexpression zu vermeiden, sowie Sicherheitsaspekte wie Immunreaktionen und Integrationseffekte.
Gene Replacement ist nicht identisch mit klassischer Gen Therapy, auch wenn beide Ansätze darauf abzielen, genetische Ursachen von Krankheiten zu adressieren. Gene Replacement ersetzt eine defekte Kopie durch eine funktionelle Kopie und setzt oft eine stabile, ggf. langfristige Expression voraus. Klassische Gentherapie kann auch andere Strategien nutzen, etwa CRISPR-basierte Editierung, um Mutationen zu korrigieren, statt einfach zu ersetzen, oder mRNA-Ansätze, die vorübergehende Expression liefern. Beide Ansätze tragen potenzielle Vorteile, aber auch Risiken.
Zu den Vorteilen von STXBP1-Gens ersetzt-Ansätzen gehören die potenzielle Korrektur zugrunde liegender Defekte, die Wiederherstellung von Synapsenfunktionen und die Möglichkeit, Modelle zu schaffen, die Therapieeffektivität zu testen. Risiken umfassen Immunantworten gegen Vektoren, unkontrollierte oder übermäßige STXBP1-Expression, ggf. Off-Target-Effekte, langfristige Ungewissheiten zur Stabilität der Expression und ethische/regulatorische Herausforderungen bei der Anwendung im Menschen. Info:
Lee Lab Gene Therapy Development
Soo-Kyung LeeUniversity at Buffalo (UB) – Department of Biological Sciences
Was ist SINEUPs-Forschung?
SINEUP-Forschung ist eine vielversprechende, neuartige therapeutische Strategie für STXBP1-Enzephalopathie und zielt darauf ab, die SINEUPs-Technologie, die auf nicht-kodierenden RNA basiert, zu nutzen, um die Funktion des STXBP1-Gens zu korrigieren und die neuronalen Symptome von STXBP1-assoziierten Erkrankungen zu behandeln. SINEUPs-Forschung untersucht eine Klasse von molekularen Werkzeugen, die als SINEUPs (Short INterspersed Element Up-regulated) bezeichnet werden und zur gezielten Steigerung der Proteinherstellung in Zellen eingesetzt werden. SINEUPs sind lange nichtkodierende RNAs (lncRNAs), die aus zwei Domänen bestehen: einer spezifischen Bindungsdomäne, die an eine Ziel-mRNA bindet, und einer Effektor-Domäne, die die Proteinproduktion ankurbelt. Ziel der Forschung ist es, SINEUPs als therapeutische Strategie zur Erhöhung der Aktivität bei genetischen Krankheiten einzusetzen.
SINEUPs können die translationelle Aktivität von Ziel-mRNAs erhöhen, indem sie deren Bindung an Polysomen (maschinelle Ribosomenkomplexe, die die Translation durchführen) erleichtern. Dies führt zu einer gesteigerten Synthese des Zielproteins.
Der Aufbau aus einer Bindungsdomäne (BD) und einer Effektor-Domäne (ED) macht SINEUPs zu einem vielseitigen Werkzeug. Forscher können die BD austauschen, um das SINEUP an eine beliebige Ziel-mRNA anzubinden und so die Produktion des gewünschten Proteins zu erhöhen.
Die Forschung konzentriert sich auf die Entwicklung von synthetischen SINEUPs, die als therapeutisches Werkzeug zur Erhöhung der Proteinproduktion in verschiedenen Zelltypen und zur Behandlung von Krankheiten, die durch einen Mangel an bestimmten Proteinen verursacht werden (z. B. Haploinsuffizienz), eingesetzt werden könnten.
Ein zentraler Aspekt der Forschung ist die Aufklärung des genauen molekularen Mechanismus. So wurde festgestellt, dass die Aktivität von SINEUPs von bestimmten RNA-Bindungsproteinen wie PTBP1 und HNRNPK abhängt und dass Methylierung an bestimmten Stellen der RNA (z. B. mit N6-Methyladenosin) für die Funktion wesentlich ist.
Info:
Sineups: A Novel Therapeutic Strategy for STXBP1 Encephalopathy
ASO-Therapie für STXBP1?
Was ist eine ASO-Therapy?
Eine ASO-Therapie steht für "Antisense-Oligonukleotid-Therapie". Diese Therapieform nutzt kurze DNA- oder RNA-Moleküle, die als Antisense-Oligonukleotide (DNA ähnliche Moleküle) bezeichnet werden, um gezielt die Expression von bestimmten Genen zu beeinflussen. ASOs (Antisense-Oligonukleotide) sind kurze, synthetische Nukleinsäurestränge. Sie können so gestaltet werden, dass sie spezifisch an die mRNA (messenger RNA) eines bestimmten Gens binden. Durch die Paarung mit der Ziel-RNA können sie die Genexpression modulieren, ohne die DNA zu verändern. Dadurch wird die Produktion des entsprechenden Proteins verändert. ASOs werden eingesetzt, um die Produktion krankheitsverursachender Proteine zu reduzieren oder zu modifizieren, und finden Anwendung bei einer Reihe von genetischen Erkrankungen.
Wenn ein ASO an die mRNA bindet, kann es auch für das STXBP1-Protein verschiedene Effekte haben. Die kurzen synthetischen Nukleinsäureketten binden gezielt an bestimmte RNA-Abschnitte im Zellkern und verhindern durch Hemmung der Transkription oder Translation die Bildung schädlicher Proteine. Durch Bindung können ASOs auch Fehl-Spleißen korrigieren oder zelluläre Abbaumechanismen wie RNA-Degradation rekrutieren.
Die ASO-Therapie zielt darauf ab, die Expression des Syntaxin-Proteins 1 zu erhöhen, indem sie beispielsweise eine hemmende mRNA blockiert oder den Abbau einer stabilen mRNA verhindert, um ein Protein zum Funktionieren zu bringen, damit Nachrichten wie gewohnt über Neuronen gesendet werden können. Theoretisch könnten ASOs drei Hauptziele verfolgen: Reduktion eines pathologisch überexprimierten oder missgefalteten STXBP1-Transkripts, Korrektur von Spleißvarianten, um eine funktionelle Isoform zu fördern, oder Modulation von Begleitwegen, um verbleibende Funktion zu erhöhen.
ASOs werden hauptsächlich bei seltenen Krankheiten eingesetzt. Sie erfordern eine sorgfältige Verabreichung, chemische Modifikationen für Stabilität und Verträglichkeit sowie eine gründliche Bewertung von Off-Target-Effekten und der langfristigen Sicherheit. ASOs wirken aber nur vorübergehend und müssen regelmäßig verabreicht werden, um eine anhaltende therapeutische Wirkung zu erreichen.
Auch wenn ASOs bei der Behandlung von Syndromen wie spinaler Muskelatrophie erfolgreich waren, können sie die BHS (Blut-Hirn-Schranke) nicht immer überwinden, sodass eine intrathekale Injektion, also eine Injektion in das Rückenmark, eingesetzt werden kann. Wenn ASOs außerdem auf das falsche Gen abzielen, besteht die Möglichkeit von Nebenwirkungen.
In der Praxis existiert derzeit keine klinisch etablierte ASO-Therapie speziell für STXBP1-Mutationen. Die Entwicklung solcher Therapien befindet sich überwiegend in präklinischen Phasen oder in frühen klinischen Studien, mit signifikantem Aufwand für Sicherheit, Wirksamkeit und regulatorische Genehmigungen. Wichtige wissenschaftliche Einschränkungen: ASOs erfordern klare Zielregionen in der mRNA, effektive Geweberekrutierung, stabile chemische Modifikationen zur Erhöhung der Halbwertszeit und Minimierung von Off-Target-Effekten. Die Wirksamkeit hängt stark von Spleißmustern, Transkriptvarianten und der Gewebebioverfügbarkeit ab. Zukünftige Perspektiven liegen in patientenspezifischen Modellen, zellbasierten Systemen (z. B. induzierte pluripotente Stammzellen) und in klinischen Studien, die Sicherheit, Dosierung und Langzeitwirkungen evaluieren. Info:
ASOs in der Forschung
Arginin-Read-Through-Sequenzierungstherapie für STXBP1?
Was ist Arginin-Read-Through-Sequenzierung?
Die Verwendung von Suppressor-tRNAs, spezialisierten Transfer-RNAs, ist ein innovativer Ansatz in der Gentherapie zur Behandlung von Krankheiten, die durch Nonsense-Mutationen verursacht werden. Die als Arginin-Read-Through-Sequenzierung (ARTS) bekannte Technik untersucht das Phänomen der Read-Through-Translation an vorzeitigen Stoppcodons in der mRNA. Arginin (eine Art Aminosäure) stabilisiert den Ribosom-mRNA-Komplex und erleichtert den Einbau von körpernahen tRNAs am Stoppcodon, wodurch das Ribosom die mRNA weiter über das vorzeitige Stoppcodon hinaus übersetzen kann. Arginin ist häufig die Aminosäure, die eingebaut wird, wenn ein UGA-Stoppcodon durchgelesen wird, was zu dem Begriff "Arginin-Read-Through" geführt hat. Das Ziel ist, ein vollständiges oder größtenteils funktionsfähiges Protein trotz des Mutations-Stoppcodons zu erzeugen.
Dieser Prozess ermöglicht die Anfügung von Aminosäuren an die wachsende Polypeptidkette, wo normalerweise ein Abbruchsignal vorhanden wäre. Das Ergebnis ist die Produktion eines Proteins in voller Länge anstelle eines verkürzten Proteins. Das Durchlesen kann durch verschiedene Faktoren beeinflusst werden, darunter der Sequenzkontext, der das Stoppcodon umgibt, und das Vorhandensein spezifischer Verbindungen, die diesen Prozess fördern. Bestimmte Aminosäuren, darunter Arginin, können das Verhalten der Ribosomen während der Translation beeinflussen. Die Effizienz des Read-through ist stark kontextabhängig und variiert je nach Stoppcodon (UAA, UAG, UGA), benachbarten Sequenzen und Gewebe sowie der Zelle, in der die Translation stattfindet. Arginnin-Read-through kann theoretisch bei Mutationen eingesetzt werden, die ein vorzeitiges Stoppkodon – nonsense-Mmutation – in einem Gen verursachen.
Arginin-Read-Through-STXBP1-Nonsense-Therapien beziehen sich auf experimentelle Behandlungen für STXBP1-Genstörungen, insbesondere solche, die durch Nonsense-Mutationen verursacht werden. Dabei wird versucht, durch die Gabe von Arginin (und möglicherweise anderen kleinen Molekülen) den Proteinbildungsmechanismus der Zelle dazu zu bringen, vorzeitige Stoppsignale (Nonsense-Codons) in der STXBP1-Boten-RNA zu "überspringen", sodass ein vollständiges, funktionsfähiges STXBP1-Protein anstelle eines verkürzten, fehlerhaften Proteins, wodurch das Kernproblem der Haploinsuffizienz bei STXBP1-RD angegangen wird. Info:
Targeting Nonsense Mutations in Diseases with Translational Read-Through-Inducing Drugs (TRIDs)
Was ist Drug Repurposing?
Drug Repurposing, auch Drug Repositioning genannt, bezeichnet die Untersuchung bereits zugelassener oder zuvor getesteter Medikamente auf neue medizinische Indikationen jenseits ihres ursprünglichen Einsatzgebiets. Statt von Grund auf neue Wirkstoffe zu entwickeln, nutzt man bestehende Arzneimittel, deren Sicherheit, Pharmacokinetik und Produktionsprozesse gut charakterisiert sind. In der aktuellen Forschung gewinnt dieses Vorgehen zunehmend an Bedeutung, weil es potenziell Kosten, Zeit und Risiko im Vergleich zur klassischen Neuentwicklung senkt. Vorteile liegen vor allem in der verkürzten Entwicklungszeit, da bereits klinische Sicherheitsdaten vorliegen, geringeren finanziellen Aufwand, eine höhere Erfolgswahrscheinlichkeit in frühen Phasen und die Möglichkeit, schnelle translationalen Schritte in klinischen Studien zu ermöglichen. Zudem können etablierte Medikamente oft rasch in etablierte Behandlungsprotokolle integriert werden, und sie eröffnen Chancen für seltene Erkrankungen, die von der ursprünglichen Indikation abweichen.
Beispiele für Drug Repurposing reichen von bewährten Medikamenten, die unerwartete Wirkungen entfalten, bis hin zu systematischen, integrierten Ansätzen, die große Datenmengen aus Genomik, Proteomik, Elektronischen Gesundheitsakten und medizinischer Literatur nutzen. Typische Fallbeispiele für STXBP1 sind 4-Phenylbutyrat, Tudcabil und Fenfluramin.
Es gibt somit generelle Ansätze, bei STXBP1-bedingten Störungen repurposed Medikamente zu prüfen, z. B. solche, die allgemeine neuronale Funktion, Synapsenstabilität, Neuroprotektion oder Metabolismus unterstützen, oder solche, die modulieren, wie Nervensysteme auf Stress reagieren. Diese Überlegungen befinden sich meist im präklinischen oder explorativen Forschungsstadium und sind noch nicht alle als bestätigte Therapien für STXBP1-Mutationen etabliert. Info:
ENDD has partnered with Panorama Medicine to repurpose FDA-approved compounds for both STXBP1
Stammzellen für STXBP1?
Was sind Stammzellen?
Stammzellen sind undifferenzierte Zellen, die sich selbst erneuern und zu spezialisierten Zelltypen differenzieren können.
Sie können aus verschiedenen Quellen gewonnen werden, darunter Knochenmark, Fettgewebe und sogar Nabelschnurblut. Diese Zellen können dann im Labor so manipuliert werden, dass sie sich zu dem gewünschten Zelltyp (z. B. Herzmuskelzellen, Nervenzellen) spezialisieren, und in den Körper des Patienten transplantiert werden, um beschädigte Zellen zu ersetzen.
Es gibt verschiedene Arten von Stammzellen.
Embryonale Stammzellen stammen aus der inneren Zellmasse einer Blastozyste und haben das Potenzial, sich in jeden Zelltyp des Körpers zu differenzieren (pluripotent).
Adulte Stammzellen kommen in verschiedenen Geweben vor und können sich in eine begrenzte Anzahl von Zelltypen differenzieren (multipotent).
Induzierte pluripotente Stammzellen (iPSCs) sind adulte Zellen, die in einen embryonalen stammzellähnlichen Zustand umprogrammiert wurden, wodurch sie sich in verschiedene Zelltypen differenzieren können.
Was ist eine Stammzelltherapie?
Die Stammzelltherapie, auch als regenerative Medizin bekannt, ist eine Behandlung, bei der Stammzellen oder deren Derivate zur Reparatur oder zum Ersatz von beschädigten oder erkrankten Geweben und Zellen eingesetzt werden. Sie zielt darauf ab, die natürliche Heilungsreaktion des Körpers zu fördern, indem Stammzellen, die sich in verschiedene Zelltypen differenzieren können, in den betroffenen Bereich eingebracht werden. Dies kann potenziell zur Regeneration von beschädigten Geweben und Organen führen und bietet einen neuen Ansatz für die Behandlung einer Vielzahl von Erkrankungen.
Ein Team von Forschern aus sechs niederländischen Wissensinstitutionen (darunter der Koordinator Vrije Universiteit Amsterdam) führt neue Forschungsmethoden ein, um die Behandlung von Entwicklungsstörungen des Gehirns, einschließlich STXBP1-bedingter Störungen, zu verbessern. Das BRAINmodel-Konsortium wird neue stammzellbasierte Methoden implementieren, um die Diagnostik und Therapieentscheidungen zu verbessern und neue Therapien zu entwickeln.
Der neue Ansatz von BRAINmodel basiert auf patienteneigenen Zellen, mit denen Netzwerke lebender Nervenzellen in einer Kulturschale hergestellt werden. Dies wird als "pluripotente Stammzelltechnologie (iPSC)" bezeichnet und bietet neue Möglichkeiten für das Verständnis menschlicher Krankheiten und die Suche nach personalisierten Behandlungen. Die Forscher werden sich mit den ethischen und sozialen Fragen befassen, die mit dem Einsatz dieser Technologien verbunden sind, die eine groß angelegte Arzneimittelforschung, einschließlich der Entwicklung neuer Medikamente, ermöglichen werden.
Das BRAINmodel-Konsortium wurde von Professor Matthijs Verhage (Vrije Universiteit Amsterdam und Amsterdam UMC, Vorsitzender) und Nael Nadif Kasri (Radboudumc, stellvertretender Vorsitzender) gegründet. Info:
BRAINmodel Consortium aims to provide better treatment for neurodevelopmental disorders
Fenfluramin für STXBP1?
Fenfluramin – FENDEEP Study – Spanien
Dies ist eine nicht kontrollierte klinische Pilotstudie (Phase 4) mit Fenfluramin als Zusatztherapie zur Behandlung von fünf verschiedenen Arten von Entwicklungs- und epileptischen Enzephalopathien (DEEs), darunter STXBP1-Enzephalopathie, mit Schwerpunkt auf epileptischen und "nicht-epileptischen Ergebnissen" bei Patienten im Alter von 2 bis 35 Jahren.
DEEs sind Entwicklungsbedingte und epileptische Enzephalopathien (schwere Epilepsieformen bei Kindern), die typischerweise im Säuglings- oder Kindesalter beginnen. Sie werden durch Gendefekte verursacht und die beeinträchtigen die Gehirnentwicklung.
Fenfluramin ist ein Medikament, das ursprünglich als Appetitzügler bekannt war, aber wegen schwerer Herzprobleme vom Markt genommen wurde, heute jedoch eine Renaissance als Antiepileptikum erlebt, hauptsächlich zur Behandlung von Anfällen bei seltenen Epilepsieformen wie dem Dravet-Syndrom und dem Lennox-Gastaut-Syndrom, indem es den Serotoninspiegel im Gehirn erhöht und so die Anfallshäufigkeit senkt. Es wird als Orphan Drug für diese seltenen Leiden zugelassen und ist unter dem Handelsnamen Fintepla erhältlich, wobei seine Anwendung streng überwacht wird.
Da STXBP1-Epilepsien oft therapieresistent sind, wird Fenfluramin auch in klinischen Studien oder im Off-Label-Use bei verwandten Syndromen untersucht, die ähnliche Merkmale aufweisen. Auch wenn STXBP1 nicht als primäre Indikation genannt wird, sind die zugrundeliegenden neurologischen Mechanismen und die Notwendigkeit für neue Therapien ähnlich. Patienten werden zurzeit nur in Madrid, Spanien, rekrutiert. Info:
Fenfluramin – klinische Studie
Tudcabil für STXBP1?
TUDCA (Tauroursodeoxycholsäure) oder Tudcabil ist ein hervorragendes Beispiel für die Umwidmung von Arzneimitteln (drug repurposing), da es sich um eine natürlich vorkommende Gallensäure handelt, die bei Gallensteinen eingesetzt wird, derzeit jedoch aufgrund ihrer antiapoptotischen und ER-Stress (endoplasmatischen Stress) reduzierenden Eigenschaften intensiv auf neue Anwendungsmöglichkeiten bei neurodegenerativen Erkrankungen (wie ALS, Parkinson's u.a.), zur Unterstützung der Leberfunktion und bei Stoffwechselproblemen untersucht wird.
Es handelt sich um einen Wirkstoff aus Bärengalle, der in der traditionellen chinesischen Medizin (TCM) seit jeher bei Leberproblemen eingesetzt wird und von der FDA für Leber/Gallensteine Erkrankungen zugelassen wurde. Es ist als chemisches Chaperon bekannt, das den Zellstress reduziert und Apoptose (Zelltod) bei neurodegenerativen Erkrankungen verhindern soll. TUDCA unterstützt die Erhaltung der Integrität von Zellmembranen und kann bei der Proteinfaltung und Fehlfaltungen helfen. Es wird auch berichtet, dass TUDCA mitochondriale Gesundheit und Funktion unterstützen kann. Info:
TUDCA – klinische Studie