Was ist STXBP1 Related Disorder?
Was ist STXBP1 Related Disorder? |
Das Gen STXBP1
Seit 2008 wurden STXBP1- Mutationen bei Patienten festgestellt. Bei dem STXBP1 Syndrom treten die Genmutationen zu einem großen Anteil de novo auf; die Mutation ist nicht von einem der Eltern geerbt, sondern sie ist im betroffenen Individuum neu aufgetreten, ganz spontan in einer Keimzelle, Samenzelle oder Eizelle der Eltern, oder in der befruchteten Eizelle. Das STXBP1 Syndrom ist eine monogenetische Erkrankung des Gehirns, d.h. es ist nur ein einzelnes Gen defekt, welches auf einem Autosom (Chromosom) liegt. Die Krankheit wird von einer autosomal-dominnate Genmutation ausgelöst; das auslösende Allel, d.h. wie ein Gen ein Merkmal ausprägt, ist nur einfach - auch heterozygot genannt - vorhanden. Heterozygot bedeutet, dass das Erbgut einer Zelle zwei unterschiedliche Kopien des STXBP1 Gens auf den beiden Chromosomen aufweist.
Der Mensch hat 23 Chromosom-Paare - also 46 Chromosomen:
- 2 Exemplaren von Chromosom 1, 2 Exemplaren von Chromosom 2, usw. bis Chromosom 22; das sind insgesamt 44 sogenannten Autosomen
- eine Frau hat zusätzlich 2 "X-Chromosomen" (Geschlechtschromosomen - d.h. 2 Gonosomen) und
- ein Mann hat zusätzlich ein "X-Chromosom" und ein "Y-Chromosom" (Geschlechtschromosomen - d.h. 2 Gonosomen)
Die Chromsomen bestehen aus bis zu 2 m lange DNA-Fäden, die an den Histonen und anderen Proteinen angelagert sind. Die 46 Chromosomen befinden sich in jeder Körperzelle.
Das STXBP1 Gen liegt auf dem langen Arm (q) des Chromosoms 9, Genlocus 34.11 und hat 20 Exons und erstreckt sich über 80.510 Basenparen. Exons sind DNA-Abschnitte eines Gens, die nach Transkription und Spleißen übrig bleiben und für Proteine codieren, z.B. Syntaxin bindendes Protein. Die Basen Adenin (A), Cytosin (C), Guanin (G) und Thymin (T) sind vier Bausteine der DNA, die in einer genau festgelegten Reihenfolge zu einer langen Kette verknüpft sind. Diese Kette ist der Schlüssel für die Kodierung des Proteins. Eine DNA enthält etwa 20.000 Genen.
STXBP1 kodiert für das Syntaxin-bindende Protein 1, ein essenzielles Protein für die Regulation der Vesikelfreisetzung und Neurotransmittersekretion in den synaptischen Spalt:
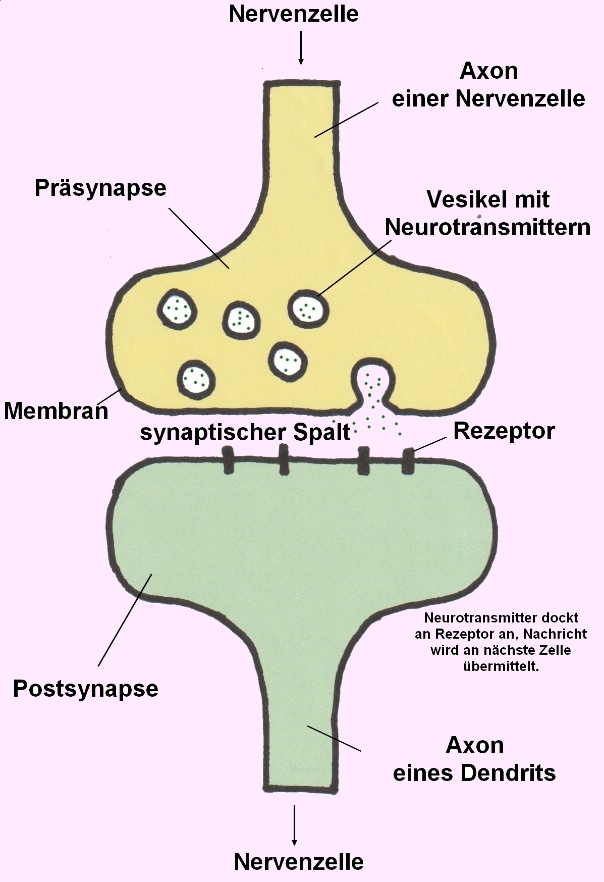
→ Prä- und Postsynapse von 2 Nervenzellen (Neuronen)- Bildnachweis: Gilberte Schnur
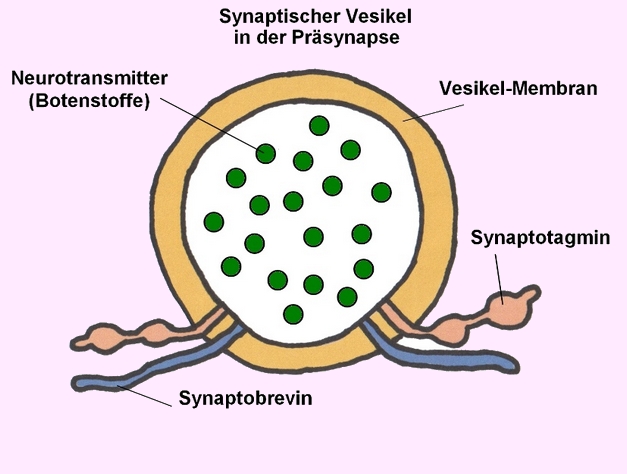
→ 1 Vesikel gefüllt mit Neurotransmittern (Botenstoffen) - Bildnachweis: Gilberte Schnur
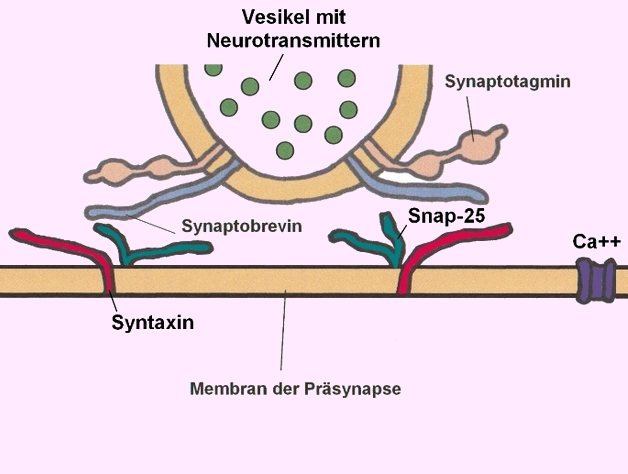
→ Membran der Präsynapse mit Syntaxin (rot) - Bildnachweis: Gilberte Schnur
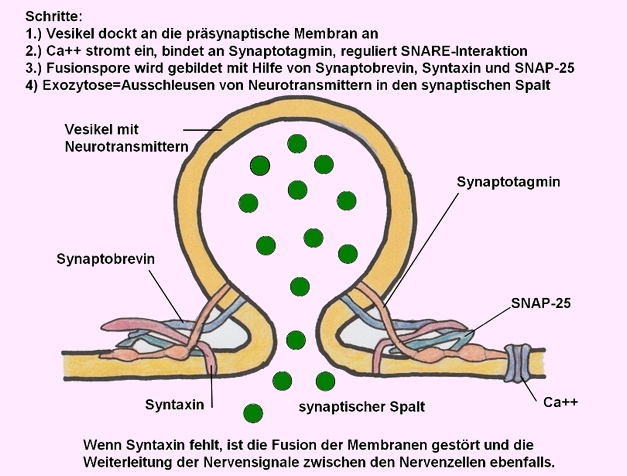
→ Ausschleusen von Neurotransmittern in den synaptischen Spalt -Funktion von Syntaxin - Bildnachweis: Gilberte Schnur
Mutationen
STXBP1 Mutationen sind Genmutationen im STXBP1 Gen selbst, sogenannte Punktmutationen. Diese können nur einzelne Basen oder auch größere DNA-Abschnitte betreffen. Bei einer Punktmutation ist nur ein Nucleotid verändert, oder nur ein Basenpaar gegen ein anderes ausgetauscht (Substitution).
Eine Substitution kann unterschiedliche Auswirkungen haben z.B.:
- stumme Mutation: es wird zwar durch den Austausch des Basenpaares ein anderes Codon gebildet, aber es wird jedoch in dieselbe Aminosäure übersetzt
- Missense-Mutation: der Austausch einer Aminosäure, deren Auswirkung auf die Funktionalität des Proteins von der Funktion des Proteins abhängt
- Nonsense-Mutation: es entsteht ein Stoppcodon, sodass die Translation vorzeitig abgebrochen wird; ein Basentriplett codiert durch Austausch eines Nucleotids zu einem Stopp-Codon
- Insertionen (Einfügen) oder Deletionen (Entfernen) einzelner Nucleotide: wenn sie im Exonbereich eines Gens auftreten, führen sie zu einer Verschiebung des Leserasters (Frameshift-Mutation), sodass ab der Läsion während der Translation die falschen Aminosäuren zur Synthese des Proteins verwendet werden.
Deletionen und Insertionen - sogenannte (Lese-)rastermutationen - beschränken sich nicht nur auf einzelne Basen, sondern können auch zwei, drei oder mehr Nucleotide umfassen.
Beispiel einer Frameshift-Mutation:
Eine Frameshift-Mutation wird durch Indels (Insertionen oder Deletionen) einer Anzahl von Nukleotiden in einer DNA-Sequenz verursacht, die nicht durch drei teilbar ist. Es verschiebt den Übersetzungsmechanismus von einem Leserahmen zu einem anderen. Nehmen wir an, wir haben eine Buchstabenfolge von THE FAT CAT SAT und der Buchstabe C wird gelöscht. Dann erhalten wir die Meldung THE FAT ATS AT ... Der Leserahmen ändert sich und das resultierende Protein funktioniert nicht richtig (oder überhaupt nicht).
STXBP1 Related Disorder - eine seltene Krankheit
Epilepsien des Kindes- und Jugendalters scheinen einen hohen genetischen Anteil zu haben, wie das auch bei dem STXBP1 Syndrom der Fall ist. Die epileptischen Enzephalopathien sind Erkrankungen bei denen eine früh einsetzende schwere Epilepsie, oft gekennzeichnet durch häufige tonische Anfälle oder Krämpfe ab dem Säuglingsalter, auftritt.
Symptome des STXBP1 Related Disorder
Die Krankheit geht mit einer Verlangsamung oder Rückbildung der Entwicklung und einer schweren kognitivsprachlichen
Entwicklungsstörung, mit schweren Bewegungsauffälligkeiten und mentaler Retardierung und auch
Verhaltensstörungen einher. Bei manchen Kindern zeigen sich auch Tremor und eine Störung der Grobmotorik / Koordination
bzw. eine Ataxie auf. Bei manchen Kindern zeigt sich auch eine schlechte visuelle Verfolgung (CVI). Es werden aber auch STXBP1-Mutation diagnostiziert, die auch weniger schwere Epilepsie-Verlaufsformen zeigen oder bei denen die Kinder keine Epilepsie entwickeln.
Bei STXBP1-RD kommt CVI (kortikale Sehbeeinträchtigung) relativ häufig vor. Studien haben ergeben, dass mehrere Patienten davon betroffen sind. Diese Krankheit bringt gravierende Entwicklungsbeeinträchtigungen mit sich und wird oft noch immer zu selten diagnostiziert. Obwohl die Augen der Patienten strukturell gesund sind, haben sie häufig Sehprobleme. Für die Diagnose von CVI sind eine gründliche augenärztliche Untersuchung sowie spezielle Untersuchungsverfahren wie VEP notwendig. Für die visuelle Rehabilitation ist es entscheidend, dass eine frühzeitige Erkennung erfolgt.
Bei STXBP1-RD wird häufig ASD (Autismus-Spektrum-Störung) oder das Vorhandensein autistischer Merkmale festgestellt. Bei Patienten sind oft sich wiederholende und aggressive Verhaltensweisen zu beobachten. Zur Diagnose von ASD werden neben Gentests, EEGs und MRTs auch weitere Instrumente wie CARS-2 (Childhood Autism Rating Scale) und ADOS-2 (Autism Diagnostic Observation Schedule) verwendet. Es ist für Kinder mit ASD wichtig, dass schon vor ihrem dritten Geburtstag interveniert wird. In dieser Phase ist das Gehirn sehr anpassungsfähig, was hilft, die langfristigen Ergebnisse zu verbessern.
Bei Patienten mit STXBP1-RD treten oft Verhaltensauffälligkeiten wie Hyperaktivität und Unaufmerksamkeit auf. Ein Verhalten, das ADHS ähnelt, wird als Teil des umfassenderen neurodevelopmentalen und verhaltensbezogenen Spektrums von STUBP1-RD anerkannt. Es ist nicht immer offensichtlich, ob Verhaltensauffälligkeiten bei STXBP1-RD auf ASD oder ADHS zurückzuführen sind.
Therapie?
Zur Zeit gibt es keine Heilung. Die Epilepsie ist bei betroffenen Kindern oft sehr schwer mit wirksamen Medikamenten einzustellen. Die Kinder haben viele Therapien wie Logo/Sprachtherapie, Physiotherapie, Ergotherapie und Sehfrühförderung, Hippotherapie.
Stxbp1 Forschungen haben angefangen, aber Forschung ist sehr teuer und auf Spenden angewiesen.